DOI:
10.1039/C5SC02395E
(Edge Article)
Chem. Sci., 2016,
7, 358-369
A bio-inspired synthesis of oxindoles by catalytic aerobic dual C–H functionalization of phenols†
Received
3rd July 2015
, Accepted 5th October 2015
First published on 6th October 2015
Abstract
Nitrogen-containing heterocycles are fundamentally important to the function of pharmaceuticals, agrochemicals and materials. Herein, we report a bio-inspired approach to the synthesis of oxindoles, which couples the energetic requirements of dehydrogenative C–N bond formation to the reduction of molecular oxygen (O2). Our method is inspired by the biosynthesis of melanin pigments (melanogenesis), but diverges from the biosynthetic polymerization. Mechanistic analysis reveals the involvement of CuII-semiquinone radical intermediates, which enable dehydrogenative carbon–heteroatom bond formation that avoids a catechol/quinone redox couple. This mitagates the deleterious polarity reversal that results from phenolic dearomatization, and enables a high-yielding phenolic C–H functionalization under catalytic aerobic conditions. Our work highlights the broad synthetic utility and efficiency of forming C–N bonds via a catalytic aerobic dearomatization of phenols, which is currently an underdeveloped transformation.
Introduction
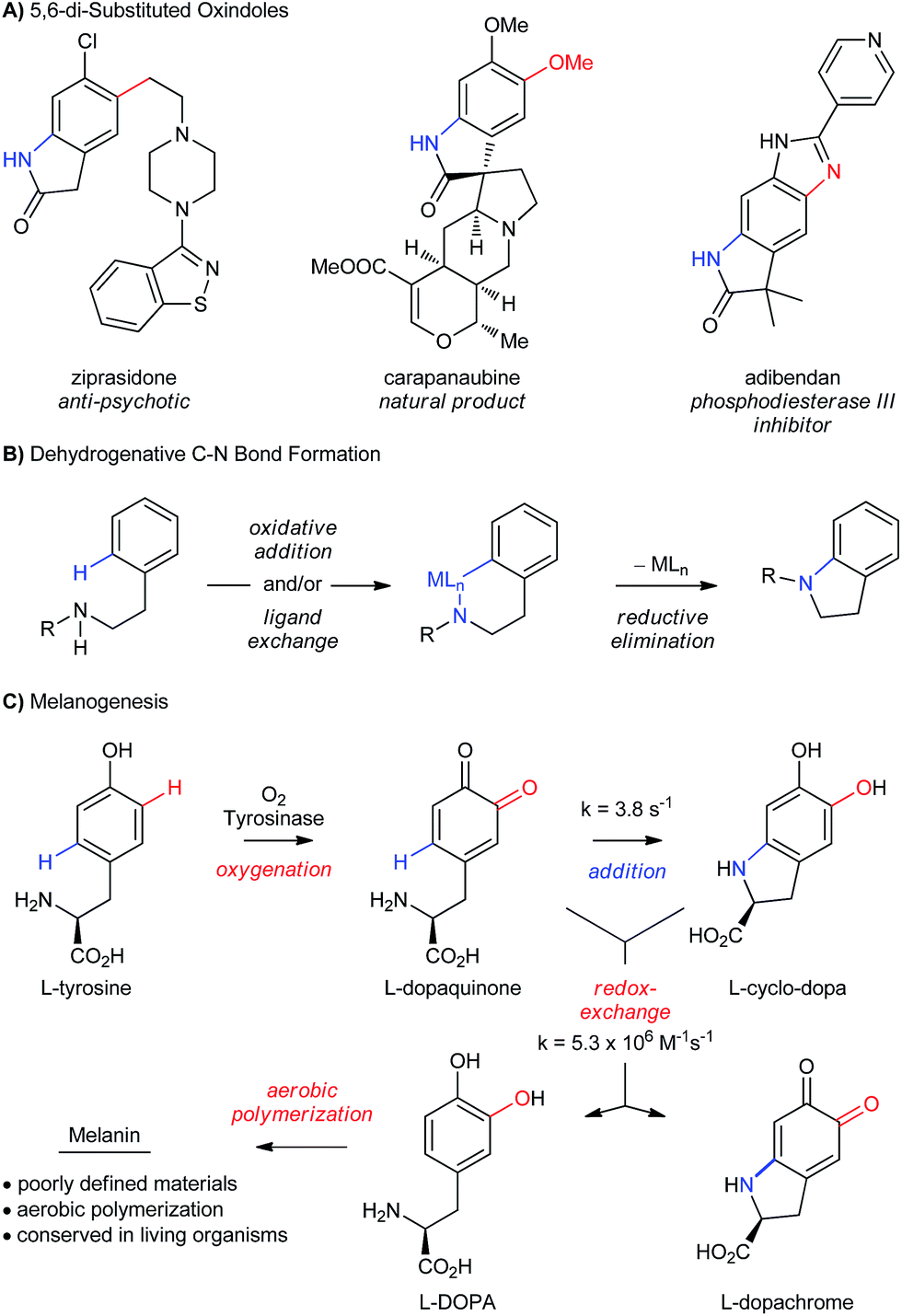
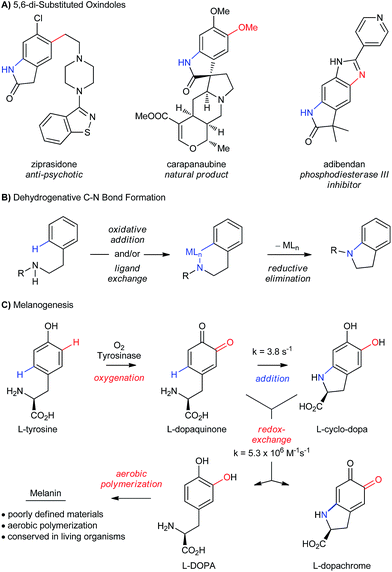
The selective oxidation of aromatic C–H bonds is critically important for the valorization of feedstock chemicals, since heteroatoms impart many desirable properties to small molecules and materials.1 Functional molecules generally contain more than one heteroatom–carbon bond (Scheme 1A), but the overwhelming majority of C–H oxidations functionalize just one bond at a time (Scheme 1B).2,3 Even with their increasing sophistication, C–H functionalizations used in sequence4 negatively impacts synthetic efficiency,5 which is compounded by increasing challenges of chemoselectivity. Thus, existing strategies to install the C–N bond of 5,6-di-substituted oxindoles (Scheme 1A) by dehydrogenative coupling would require selective functionalization of the product at C5 and C6, or would need to be chemoselective to existing functionalities at these positions.
 |
| | Scheme 1 (A) Biologically active 4,5-disubstituted oxindoles. (B) Traditional C–N bond formation by cross-coupling. (C) Proposed mechanism for the biosynthesis of melanin pigments. | |
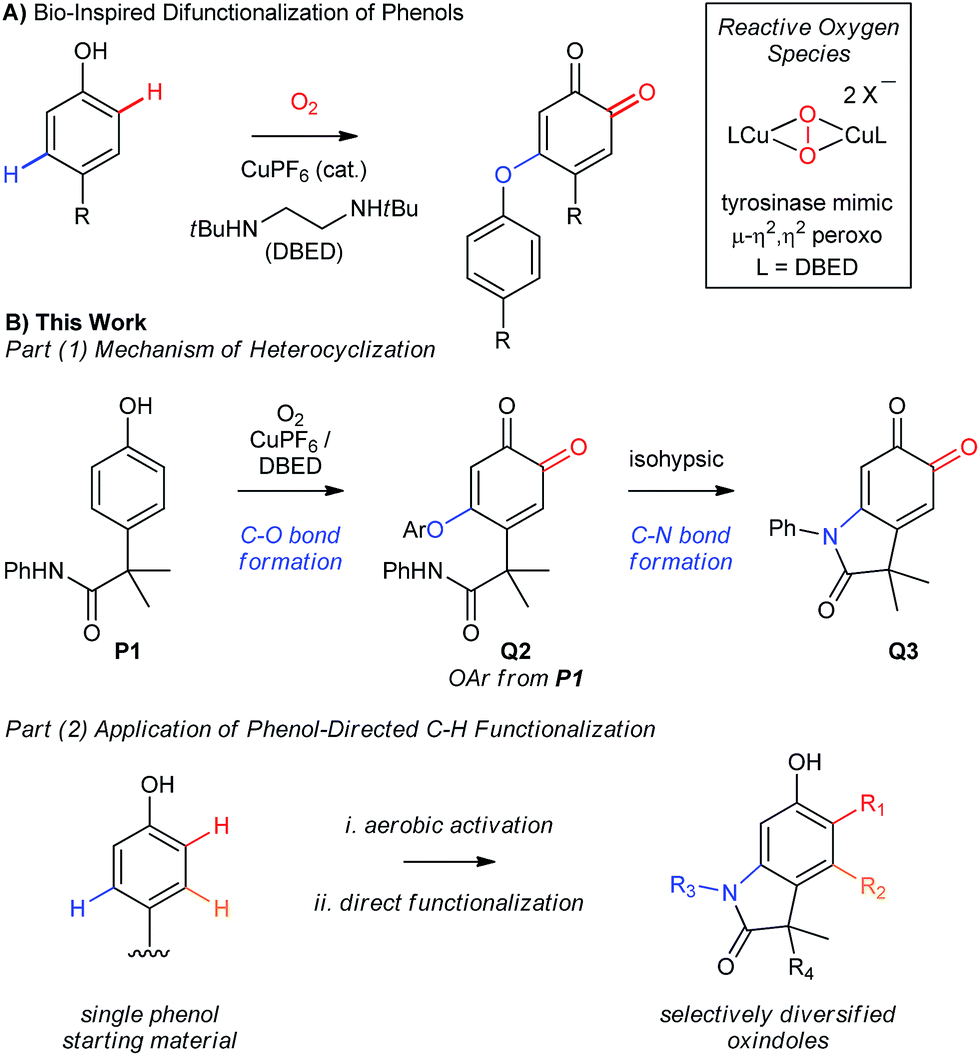
An alternative approach to aromatic C–H functionalization occurs during the biosynthesis of melanin pigments,6 whereby the aromatic C–O and C–N bonds of L-cyclodopa are installed by a phenol-directed dual C–H functionalization (Scheme 1C). Such high levels of efficiency and simplicity for dehydrogenative heteroatom–carbon bond formation are attractive,2a,7–9 but melanin is a complex, irregular bio-material, whose heterogeneity reflects an inherent complication of installing heteroatoms by this mechanism. L-Cyclodopa is significantly more electron-rich than L-tyrosine, and its oxidation to L-dopachrome is facile. This can occur by autoxidation or by redox exchange with L-dopaquinone, which is competitive with C–N cyclization at millimolar concentrations (Scheme 1C).10 This affords a complex mixture of redox-active intermediates that is ultimately translated into the bio-material. In a laboratory setting, this contributes to poor selectivity, which is a well-known challenge when oxidizing phenols with O2.11 With the exception of Patureau's recent work12 and Hay's industrial aerobic polymerization of 2,6-dimethyl phenol,13 functionalizing the C–H bonds of phenols with heteroatoms requires stoichiometric quantities of an external oxidant11a,14,15 or oxidation of the heteroatom prior to coupling.16 And while the merits of developing catalytic aerobic alternatives are clear,17 progress has been slow in the case of phenols, due in part to their facile oxidation to phenoxyl radicals.11,18
The potential efficiency of a controlled “melanogenic” functionalization inspired one of our groups to develop a catalytic aerobic transformation that uses a small-molecule mimic of the enzyme tyrosinase (Scheme 2A).19 Tyrosinase is a type-III Cu-enzyme responsible for triggering melanogenesis.11b,20 It avoids radical-based oxidations of phenols by confining O2-activation and oxygen atom transfer to the inner coordination sphere of its dinuclear Cu active site. This is a well-accepted strategy for avoiding radical oxidations,11c,18,21 but it remains difficult to implement in the absence of the protein matrix.11b Inspired by the work of Stack22 and others,23 we developed conditions for the selective ortho-oxygenation of phenols that employ catalytic amounts of [CuI(CH3CN)4](PF6) (abbreviated CuPF6), N,N′-di-tert-butylethylenediamine (DBED) and O2 at room temperature.19,24,25 Unlike the enzyme, however, which is selective for ortho-oxygenation, our conditions return coupled ortho-quinones that have undergone an additional C–H bond oxidation. This highlights important differences between the mechanism of our catalytic transformation and the mechanism of the enzyme, which we explore in the present work.
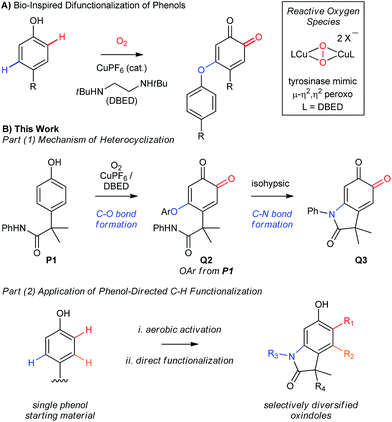 |
| | Scheme 2 (A) Previous work from our groups. (B) This work: development of a catalytic aerobic functionalization of phenols. | |
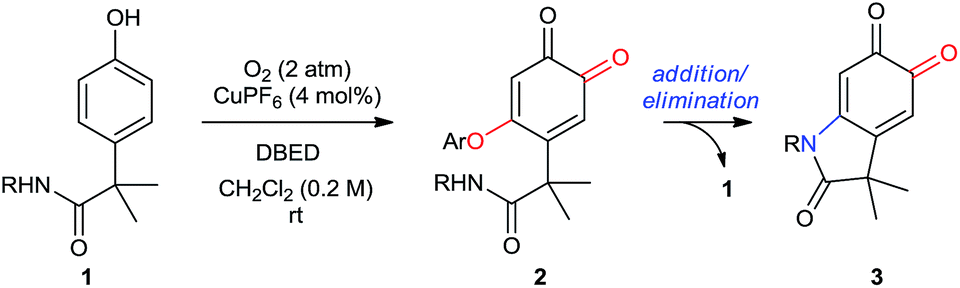
In Part 1 of this manuscript, we demonstrate that the heterocyclization of phenol P1 to oxindoloquinone Q3 occurs by a mechanism of homo-coupling and substitution, such that C–N bond formation is isohypsic5d–f (Scheme 2B). This is distinct from the commonly accepted mechanism of melanogenesis (Scheme 1C),6a and it provides important benefits to the efficiency and selectivity of heterocyclization by circumventing redox exchange. In Part 2, we apply our dual C–H functionalization to a synthesis of oxindoles, which highlights the versatility of activating phenols as their ortho-quinones.26 Unlike traditional cross-coupling reactions, which afford stable products following a single C–N coupling, the ortho-quinone obtained by our transformation is an activated precursor to the aromatic heterocycle. ortho-Quinones participate in a range of complexity-generating transformations,6a,26 allowing us to rapidly diversify the oxindole product from a single phenol starting material. This strategy is unique amongst dehydrogenative couplings, in that the energy stored in O2 is not only used to install one aromatic C–N bond, but also creates an activated product that is readily amenable to further functionalization. While this is a general feature of reactions that de-aromatize phenols,14i,27 our case marks a rare example where de-aromatization is conducted by aerobic catalysis.17,28
Results and discussion
Part 1. Mechanistic investigation
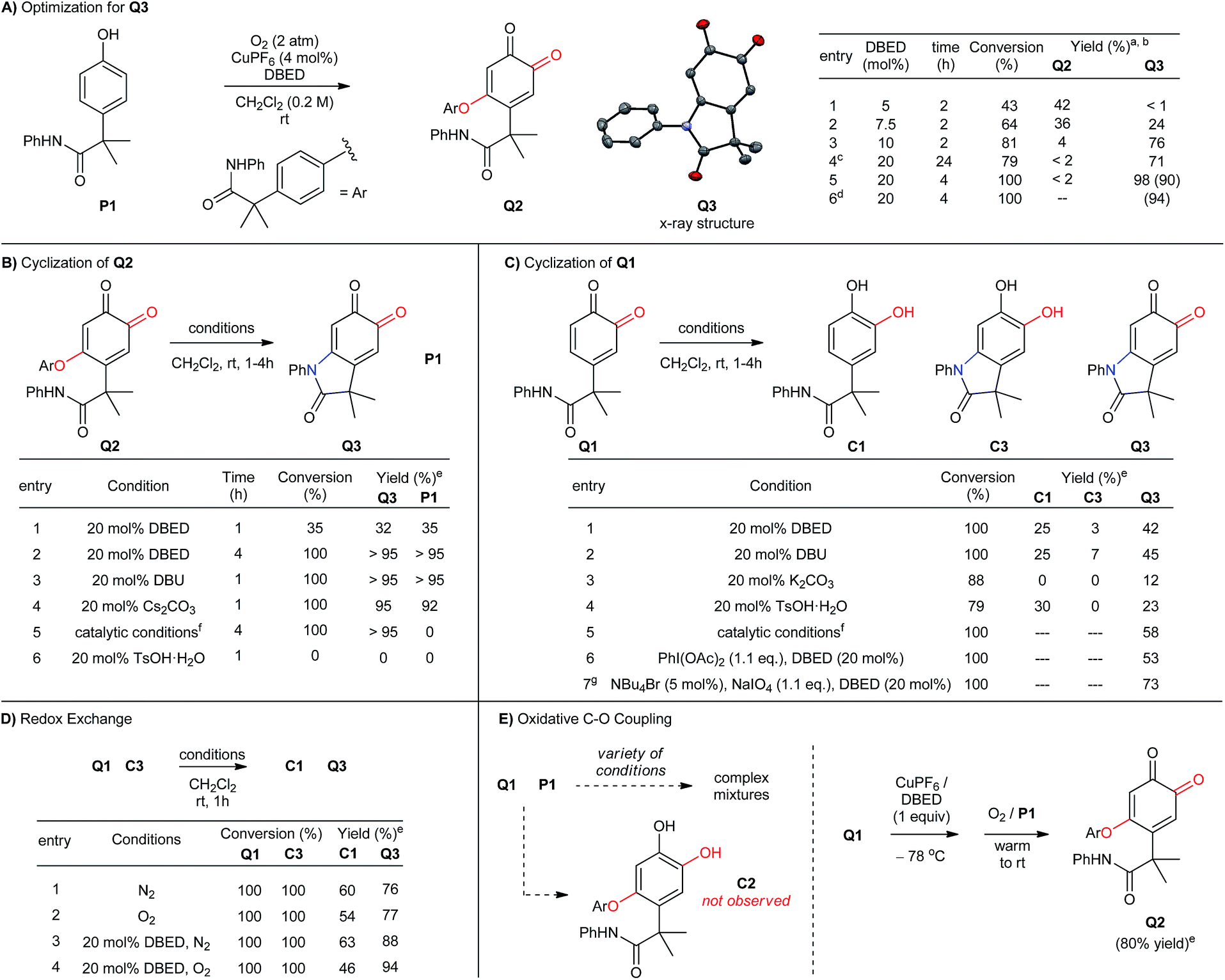
Reaction optimization and control experiments.
When we examined the oxidative functionalization of acetanilide P1 under our standard reaction conditions,19 we discovered a surprising mixture of O-coupled ortho-quinone Q2 and oxindoloquinone Q3 (Scheme 3A). The ratio of these products is sensitive to the quantity of DBED relative to CuPF6, so that a [DBED]/[Cu] ratio of 5/4 is completely selective for Q2, 7.5/4 affords a mixture and 10/4 favours Q3 after 2 h (entries 1–3). This dependence on DBED suggests a base-promoted cyclization of Q2 as a key step in the formation of Q3 (see below), which is surprising since the conventional mechanism of melanogenesis does not involve phenolic C–O coupling prior to C–N coupling. While open-flask conditions are tolerated (entry 4), incomplete conversion after prolonged reaction times led us to use an overpressure of 1 atm of pure O2 for our optimized conditions. Thus, oxygenation of P1 for 4h at room temperature in the presence of 4 mol% CuPF6 and 20 mol% DBED leads to a 90% yield of isolated oxindoloquinone Q3 at complete conversion (entry 5). Reaction efficiency is maintained on 5 g scale, wherein Q3 is isolated in 94% yield (entry 6).
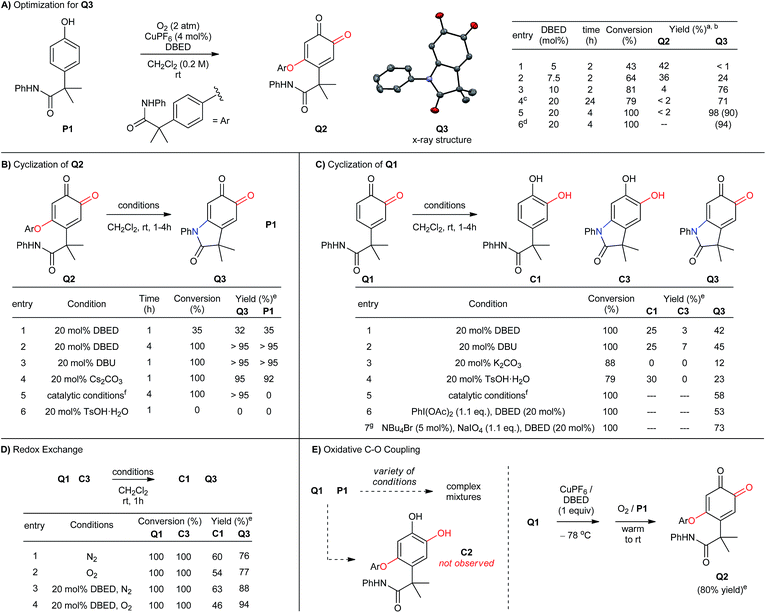 |
| | Scheme 3 (A-E) Optimization of reaction conditions and control experiments. (a) Reactions performed with 0.5 mmol of P1. Work-up: 10% aqueous NaHSO4. (b) Product yield determined by 1H-NMR using hexamethylbenzene as an internal standard. Isolated yield reported in parenthesis. (c) Reaction performed under open-flask conditions. (d) Reaction performed using 5 g of P1. (e) Reactions were performed with 0.1 mmol of starting material, and analyzed by 1H-NMR following acidic work-up using hexamethylbenzene as an internal standard. (f) Catalytic conditions: [Cu(MeCN)4](PF6) (4 mol%), DBED (20 mol%), O2 (2 atm). (g) Reaction performed in a biphasic mixture of CH2Cl2 (10 mL) and water (2 mL). | |
The cyclization of Q2 can be promoted by a variety of bases and cleanly returns Q3 and P1 (Scheme 3B, entries 1–4).29 Oxindoloquinone Q3 is the only observed product if Q2 is re-subjected to the standard catalytic conditions (entry 5), demonstrating that Q2 is a competent reaction intermediate in the transformation of P1 into Q3, and that upon its release from Q2, P1 can re-enter the catalytic cycle for conversion to Q3. Cyclization of Q2 to Q3 is not promoted by acid (entry 6), suggesting that Q2 does not cyclize to an appreciable extent during the acidic work-up.
C–N bond formation by substitution is significantly more efficient than intramolecular cyclization of the acetanilide within ortho-quinone Q1 (Scheme 3C). To evaluate this transformation, we synthesized Q1 from P1 by using Pettus' ortho-oxygenation method with IBX.16f In the absence of an oxidant, exposure of Q1 to 20 mol% DBED affords a 42% yield of Q3, along with a 25% yield of C1 and trace amounts (<5%) of C3 at complete conversion of Q1 (entry 1). Selectivity for Q3 improves if the cyclization is performed under oxidizing conditions (entries 2–4), but never as cleanly as catalytic conditions directly from P1 or base-promoted cyclization of Q2. The results of entry 1 are consistent with a redox exchange between C3 and Q1, whose feasibility was confirmed independently by mixing equimolar amounts of Q1 and C3 under a variety of conditions (Scheme 3D, entries 1–4). In each case, redox exchange is accompanied by a significant loss of mass balance (up to 69%), making the Q1-to-C3-to-Q3 pathway inconsistent with the high selectivity observed under our catalytic conditions.
The cyclization of Q2 is said to be isohypsic,5d–f in that additional oxidation is not required to arrive at target quinone Q3. This avoids the possibility of a redox exchange following C–N bond formation between Q1 and C3. While this may account for the efficiency of C–N bond formation, it cannot account for the formation of Q2, which represents an oxidative coupling between Q1 and P1 (Scheme 3E). This coupling reaction should be equally sensitive to the formation of electron-rich catechol C2, which would be capable of redox exchange with Q1. In all of our attempts to perform the addition of P1 to Q1 in the absence of Cu and O2, we observed complex reaction mixtures (see Table S5 in the ESI†). This highlights well-known difficulties of performing a nucleophilic addition to ortho-quinones via a straightforward 2-electron process.30 If, however, Q1 is premixed with equimolar quantities of CuPF6 and DBED prior to the addition of P1 under O2, Q2 is obtained in yields that range from 80 to 95% (Scheme 3E). This is consistent with a Cu-mediated dehydrogenative coupling between P1 and Q1 to afford Q2 that does not generate catechol C2 as an intermediate. Additional support for this hypothesis is provided by the complete absence of Q3 if C2, or if a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of C2 and P1, is subjected to the standard catalytic conditions. These results are surprising since catechols are considerably more electron-rich than phenols and are generally easier to oxidize to the ortho-quinone. Nevertheless, a catalyst system composed of 4% CuPF6 and 20% DBED does not promote an efficient oxidation of C2, nor of C3 (see ESI†). The absence of catechols under our catalytic conditions contrasts with the previous work of Maumy and Capdevielle, who proposed catechols as viable products of oxygenation under their stoichiometric Cu-mediated conditions (so-called “corrosion method”).31 We suspect that in both cases, the immediate product of oxygenation is a quinone–Cu complex (see below),25,32 and that the fate of this intermediate rests in the precise nature of the reaction conditions.
:1 mixture of C2 and P1, is subjected to the standard catalytic conditions. These results are surprising since catechols are considerably more electron-rich than phenols and are generally easier to oxidize to the ortho-quinone. Nevertheless, a catalyst system composed of 4% CuPF6 and 20% DBED does not promote an efficient oxidation of C2, nor of C3 (see ESI†). The absence of catechols under our catalytic conditions contrasts with the previous work of Maumy and Capdevielle, who proposed catechols as viable products of oxygenation under their stoichiometric Cu-mediated conditions (so-called “corrosion method”).31 We suspect that in both cases, the immediate product of oxygenation is a quinone–Cu complex (see below),25,32 and that the fate of this intermediate rests in the precise nature of the reaction conditions.
Spectroscopic investigation.
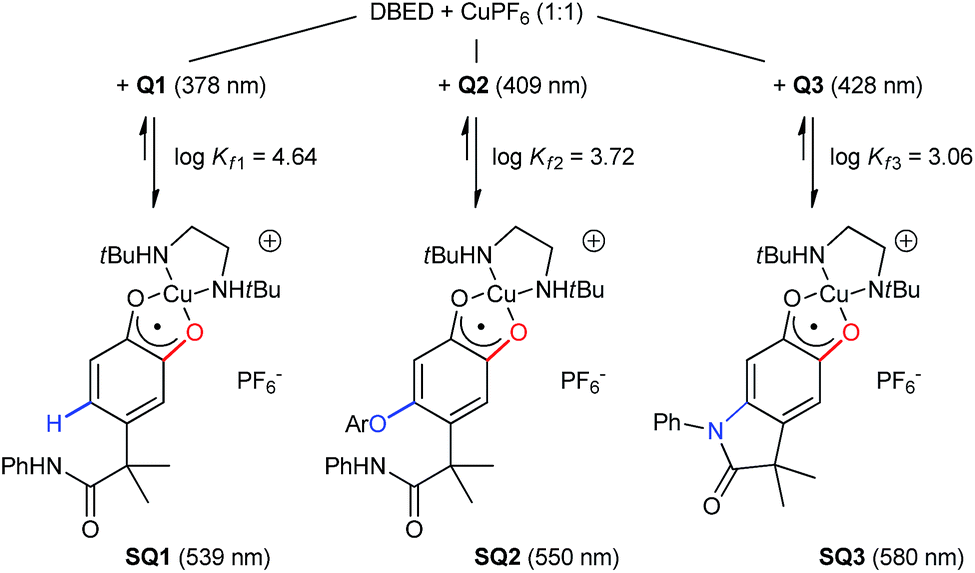
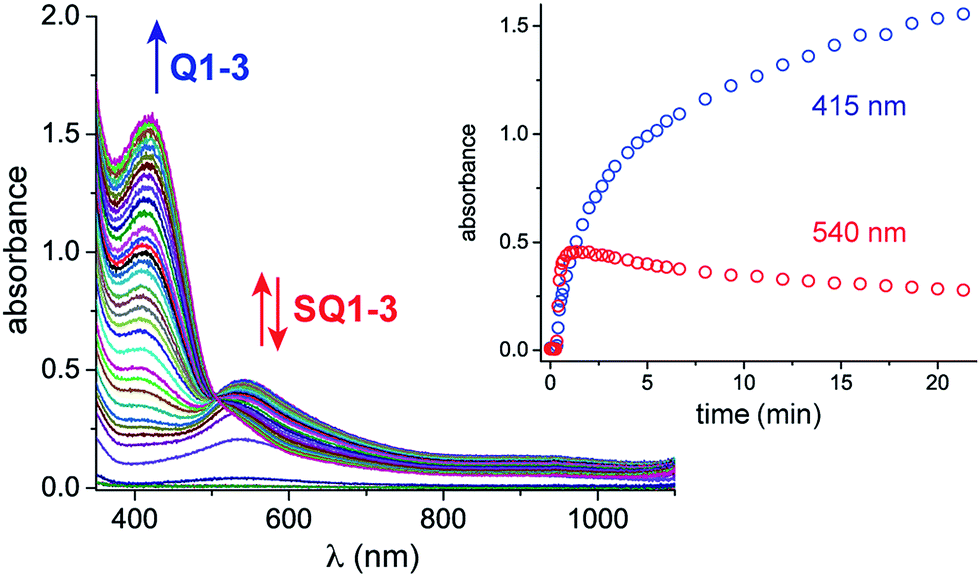
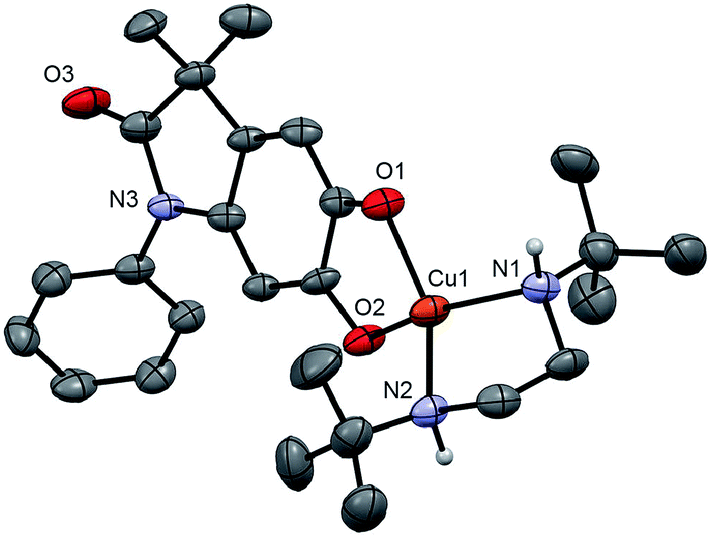
Monitoring the conversion of P1 into Q3 by in situ UV-visible spectroscopy reveals the intermediacy of Cu(II)-semiquinone radicals SQ1–3 (Scheme 4), which possess characteristic absorption bands at ∼550 nm (Fig. 1a, Fig. S6†). These structural assignments are supported by previous work from us24,25 and Stack,22 ESI-MS characterization of the reaction mixture at short reaction times (Fig. S1–S2†), and the independent synthesis of SQ1–3 from Q1–3, CuPF6 and DBED (Scheme 4). Thus, coordination of Q1–3 with DBED-Cu(I) is thermodynamically favoured, and elicits the transfer of one electron from the metal to the quinone (Scheme 4). Stronger binding is observed with the more electron-poor Q1 than Q2 or Q3, spanning more than one order of magnitude. The viability of SQ1–3 as reaction intermediates was confirmed in experiments using them as the sole source of Cu (Section 4e of the ESI†). Thus, the efficiency and selectivity in the transformation of P1 to Q3 are not affected when SQ1–3 are prepared independently, and then employed as pre-catalysts under otherwise identical conditions. The solid-state structure of SQ3, obtained by single-crystal X-ray analysis of its SbF6− salt (Fig. 2), reveals a pseudo-tetrahedral coordination environment around Cu(II) with a 50.3° dihedral angle between the NCuN and OCuO planes, and C–O bond lengths within 1.278–1.291 Å, consistent with previously reported Cu(II)-semiquinones.25,32,33 Finally, the correspondence between the electronic structures of the intensely purple SQ1, SQ2 and SQ3 (Fig. S6†) and those previously reported in the literature provides additional support for their assignment as DBED-ligated Cu(II)-semiquinone complexes.25,32
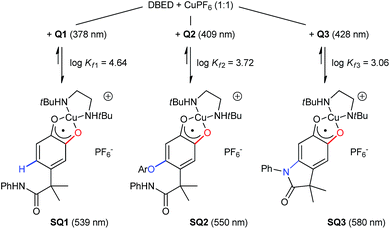 |
| | Scheme 4 Independent synthesis of SQ1–3 by coordination and electron-transfer of DBED-Cu(I) to Q1–3, with binding constants and main spectral features. Details in Fig. S3–S6.† | |
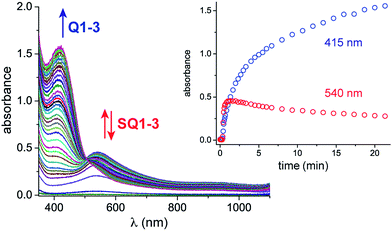 |
| | Fig. 1
In situ UV-vis spectroscopic monitoring under catalytic conditions: CH2Cl2, 25 °C, 15.67 mM P1, 9% CuPF6, 20% DBED, O2 (2 atm), 1.0 mm pathlength. Inset: absorbance profile at 415 and 540 nm. To ensure homogeneous mixtures of P1, UV-vis experiments were conducted at a low concentration of P1. To improve the rate of conversion, the Cu loading was increased to 8–9%. | |
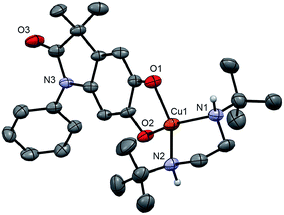 |
| | Fig. 2 ORTEP at 50% ellipsoid probability of one molecule of SQ3, a cation, in the X-ray structure of (SQ3)2(SbF6)2·2.5 CH2Cl2. H atoms except those on N1 and N2 are omitted for clarity. Selected bond lengths (Å): Cu–N1 1.992(5), Cu1–N2 1.994(4), Cu1–O1 1.955(4), Cu1–O2 1.946(4), C1–O1 1.284(6), C2–O2 1.291(6). Dihedral angle between N1Cu1N2 and O1Cu1O2 planes: 50.34°. | |
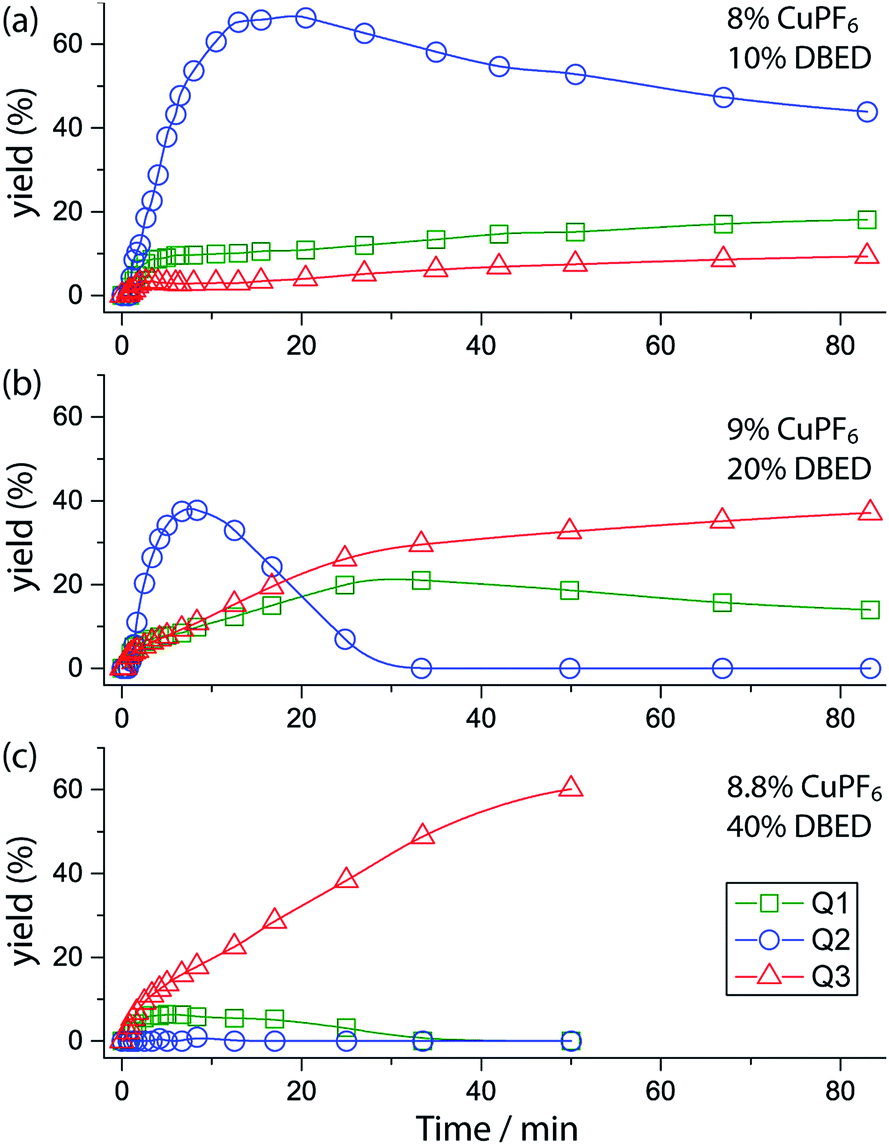
Time profiling of all visible species (Q1–Q3 and SQ1–SQ3) at different DBED/CuPF6 ratios reveals how DBED in excess of CuPF6 influences the ratio of Q2 and Q3 (Fig. 3), as well as the speciation of SQ1–3 (Fig. S7–S9†). UV-vis monitoring of the catalytic oxygenation of P1 with a small excess of DBED relative to CuPF6 (10% DBED and 8% CuPF6 per P1; 1.25 ratio) shows the fast formation of SQ1 as a ∼1:1 mixture with SQ2, which gradually converts to Q2 as the reaction progresses (Fig. 3a and S7†). Appreciable amounts of Q3 (>10%) are only observed after 1 h, consistent with the results of entry 1 in Scheme 3A, which return Q2 as the major product when the DBED/CuPF6 ratio is 1.25. When the concentration of DBED is increased to a ∼2-fold excess relative to CuPF6, SQ1–3 form rapidly, and gradually convert to Q3 over the course of 1 h (Fig. 3b and S1, S2 and S8†). Importantly, the quantity of Q2 goes through a maximum, strongly suggesting that it is an intermediate in the formation of the final product, Q3. The use of a ∼4-fold excess of DBED results in the most rapid formation of Q3, supporting our hypothesis that Q3 forms by a DBED-promoted cyclization of Q2 (Fig. 3c and S9†). A notable feature of the reaction speciation is the consistently low concentration of Q1. Of all the quinones, Q1 is the least substituted and most electron-deficient, making it the most reactive towards non-discriminant nucleophilic attack. By remaining bound to Cu as SQ1, the most prevalent semi-quinone intermediate in all experiments, Q1 is effectively protected as a partially reduced, Cu-ligated radical anion, whose stability is enhanced by steric shielding of the ligand environment.
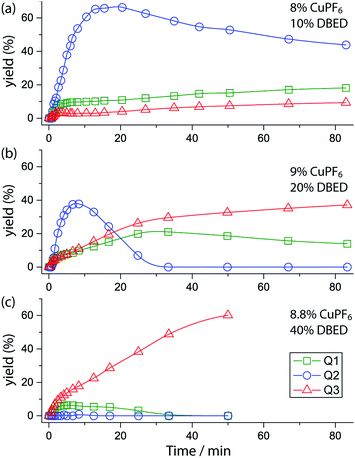 |
| | Fig. 3 Yields of Q1 (green squares), Q2 (blue circles), and Q3 (red triangles) during the reaction under three sets of conditions, deduced by fitting UV-vis spectra at various time points (see Fig. S7–S9† for full data including SQ1–3). Conditions: CH2Cl2, 25 °C CH2Cl2, O2 (1 atm), 15.67 mM P1 and (a) 8% CuPF6, 10% DBED; (b) 9% CuPF6, 20% DBED (as in Fig. 1); (c) 8.8% CuPF6, 40% DBED. The y-axis is scaled to the maximum concentration of each species, i.e. [Q1]max = [Q3]max = [P1]0 and [Q2]max = 0.5 [P1]0. Thus each point in the graph gives the yield of each species. | |
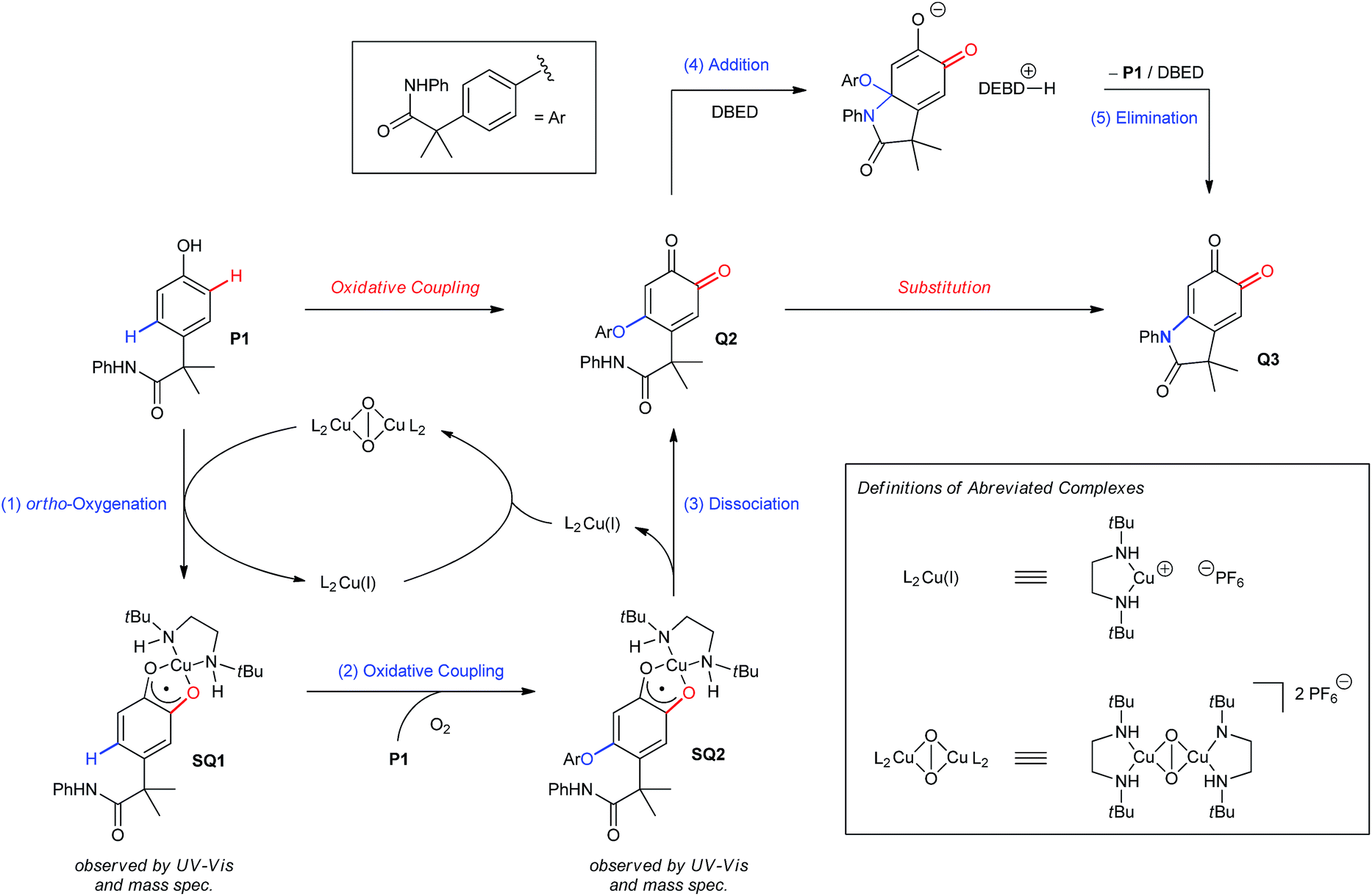
Proposed catalytic cycle.
The results from our mechanistic studies lead us to propose the catalytic cycle illustrated in Scheme 5. ortho-Oxygenation of P1 with a DBED2Cu2O2 peroxo species25 affords a mixture of Q1 and SQ1, which we observe by UV-vis and ESI-MS. The feasibility of an oxidative coupling between SQ1 and P1 in the presence of O2 was demonstrated in Scheme 3E, and results in the formation of SQ2, which is also observed by UV-vis and ESI-MS. Dissociation of SQ2 releases Cu(I) and closes the catalytic cycle of ortho-oxygenation, setting the stage for a base-promoted cyclization of Q2 to generate Q3. Addition of the pendant amide triggers the elimination of P1, which re-enters the catalytic cycle, consistent with entry 6 of Scheme 3B. Under these conditions, heteroatom–carbon bond formation is either oxidative (step 2) or isohypsic (steps 4 and 5), enabling C–H functionalization without the formation of an electron-rich catechol. This is an important feature of the transformation, since it provides a mechanistic framework for the functionalization of ortho-quinones that avoids problems of selectivity that can be associated with redox-exchange.
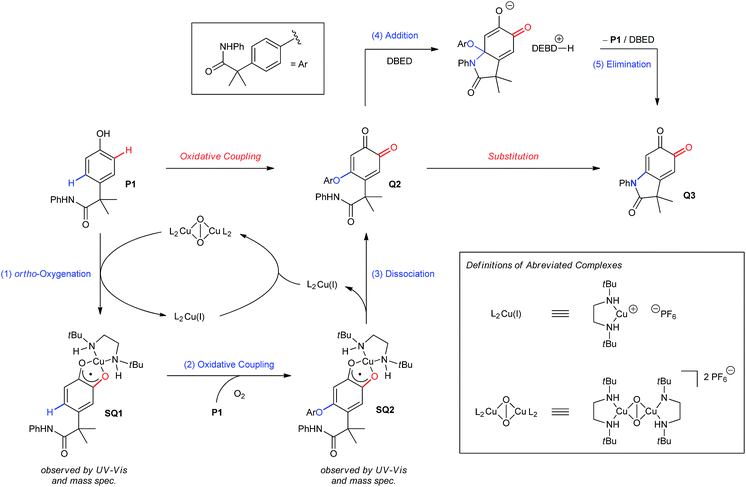 |
| | Scheme 5 Proposed mechanism. | |
The involvement of Cu beyond ortho-oxygenation creates an important distinction with melanogenesis, wherein Cu remains bound within the active site of tyrosinase.20 In melanogenesis, spontaneous cyclization of dopaquinone into cyclodopa outside of the enzyme's active site produces a quinone/catechol redox-couple that can engage in redox exchange (Scheme 1B).10 To what extent redox exchange contributes to the complexity of the melanin polymer remains unclear, but its negative impact on the cyclization of Q1 is clear (Scheme 3C). Thus, we attribute the high degrees of selectivity under our catalytic conditions to the intimate involvement of Cu during and following ortho-oxygenation, which sequesters Q1 as the SQ1 complex, and promotes an oxidative coupling with P1 that avoids the formation of an intermediate catechol, and thus redox-exchange. ortho-Quinones remain underutilized in organic synthesis, since they are largely viewed as reactive intermediates that are prone to spontaneous polymerization. We demonstrate here that many of these complications can be avoided by metal complexation, which should become a general strategy for exploiting the broad reactivity of these intermediates.
Part 2. Synthetic utility
Scope of substituents on nitrogen.
Cross-coupling reactions that form C–N bonds by reductive elimination are sensitive to the steric and electronic properties of the nitrogen atom undergoing bond formation.1a,34 This is particularly evident in dehydrogenative C–N bond forming reactions, which are typically restricted to a single substituent on nitrogen for a given set of conditions.7d,35 This is not the case under our conditions, where C–N bond formation is a base-promoted process that does not require a transition metal. As a result, a broad range of nitrogen substituents possessing very different pKa values are accommodated by making only slight adjustments to the reaction pH (Table 1). For example, when our standard conditions are applied to the cyclization of benzyl amide 1a, coupled ortho-quinone 2a is formed selectively (entry 2), due to the decreased acidity of the amide N–H bond relative to P1 (entry 1). Correspondingly, selectivity for the oxindoloquinone is restored by adding 30 mol% of the more basic 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (entry 3). Alternatively, cyclization of an O-methyl hydroxamic ether under standard conditions (entry 4) returns significant decomposition that we attribute to the increased acidity of the N–H bond and the potential lability of the N–O bond. Therefore, to restore selectivity for the oxindoloquinone, we simply decrease the amount of DBED to 5 mol% (entry 5). Finally, the successful cyclization of a Boc-imide (entry 6) highlights the breadth of substituents that are tolerated on nitrogen, which include aryl, alkyl, methoxy or carbonyl. To our knowledge, there are no other examples of a dehydrogenative amination reaction that tolerate comparably diverse substituents on the nitrogen nucleophile.2
Table 1 Scope of substituents on nitrogena
|
|
| Entry |
R |
Approximate pKa (DMSO)b |
DBED (mol%) |
Time (h) |
Conversion (%) |
Yield of 2c (%) |
Yield of 3c,d (%) |
|
Reactions performed with 0.5 mmol of 1.
These approximate pKa values for the amide N–H are derived from Bordwell's or Evans' pKa tables.
Product yield determined by 1H-NMR using hexamethylbenzene as an internal standard.
Isolated yield in parenthesis using 1 mmol of 1.
DBU (30 mol%) was used as additive.
Due to poor substrate solubility, the reaction was performed at 45 °C.
|
| 1 |
Ph (P1) |
22 |
20 |
4 |
100 |
— |
98 (90) |
| 2e |
Bn (1a) |
26 |
20 |
4 |
80 |
56 |
4 |
| 3 |
Bn (1a) |
|
20 |
8 |
78 |
<2 |
67 (61) |
| 4 |
OMe (1b) |
17 |
20 |
4 |
74 |
— |
37 |
| 5 |
OMe (1b) |
|
5 |
4 |
86 |
— |
72 (59) |
| 6f |
Boc (1c) |
18 |
20 |
4 |
93 |
— |
86 (84) |
Aryl substitution on nitrogen.
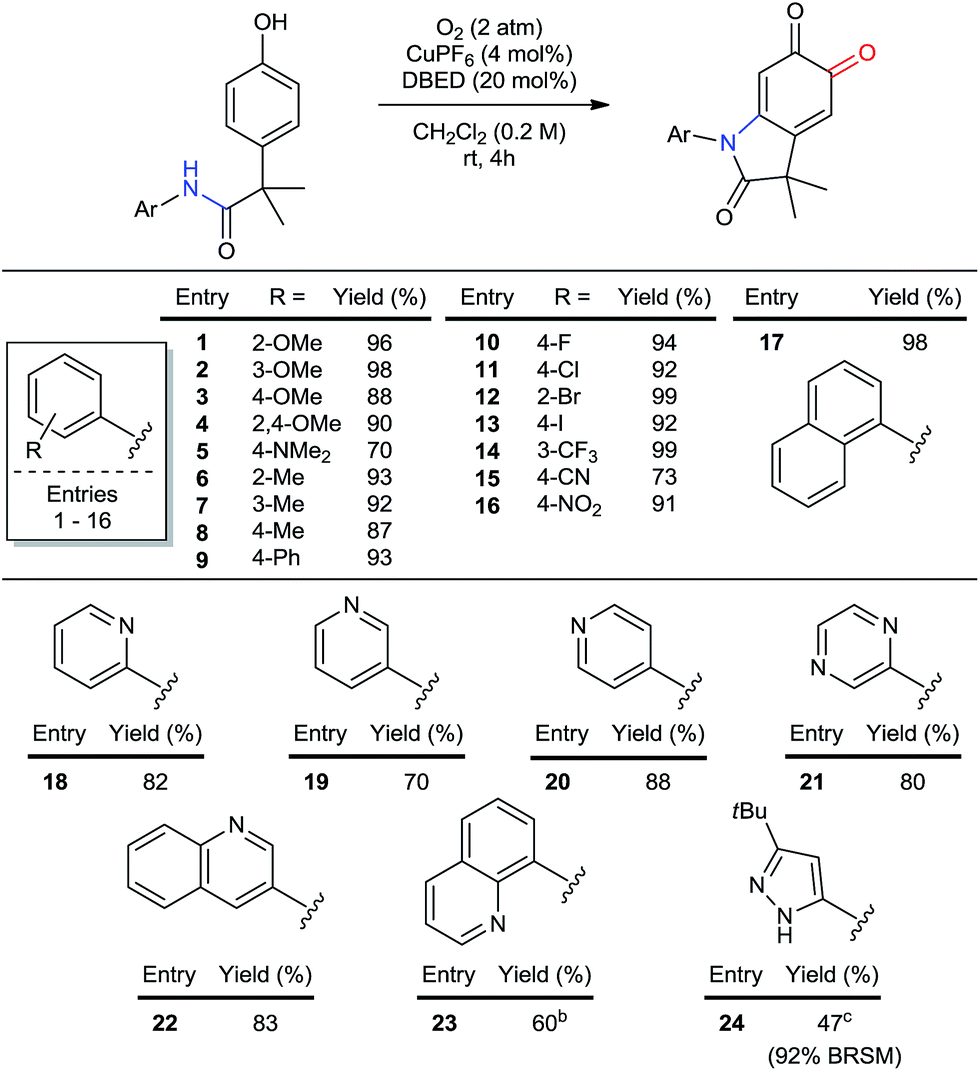
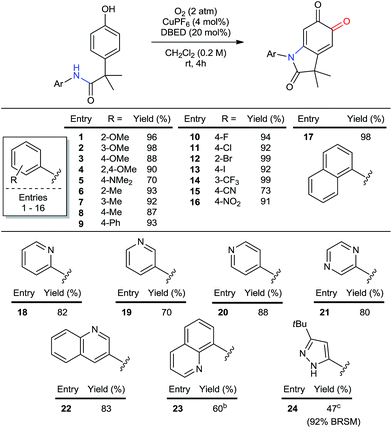
The particular importance of N-aryl36 linkages in pharmaceuticals and agrochemicals1a,b,36d prompted us to investigate the scope of aryl substitution on the acetanilide (Table 2). Both donating and withdrawing groups are tolerated on the aryl ring (entries 1–16), including the strongly electron-donating and redox-sensitive N,N-dimethylamine (entry 5). Primary benzylic hydrogen atoms are also tolerated (entries 6–8), as are halogen substituents (entries 10–14), demonstrating compatibility with C–H bonds that are susceptible to aerobic oxidation,11c,37 as well as functional groups classically employed in cross-coupling reactions. A particular advantage of using Cu to oxidize C–H bonds is its resistance to heteroatom poisoning, which frequently limits related transformations catalyzed by precious metals.38 Under our conditions, a broad range of heteroaromatic rings are tolerated, affording pyridine-, pyrazine-, pyrazole- and quinoline-substituted oxindoloquinones in good yields (entries 18 to 24). These examples also highlight the compatibility of our method with directing groups commonly used for directed C–H functionalization (entries 18 and 23),2g,39 and sets the stage for substrate diversification by orthogonal amide-directed C–H oxidations (see Scheme 7 below).
Table 2 Scope of aryl and hetero-aryl substituents on nitrogena
|
Reactions performed on 1 mmol scale and reported yields are of the purified oxindoloquinone.
30 mol% DBED, 6 h.
A 20%-by-volume mixture of THF in CH2Cl2 was used due to issues of solubility.
|
|
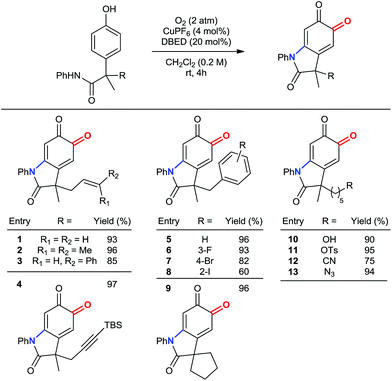
Scope of benzylic substituents.
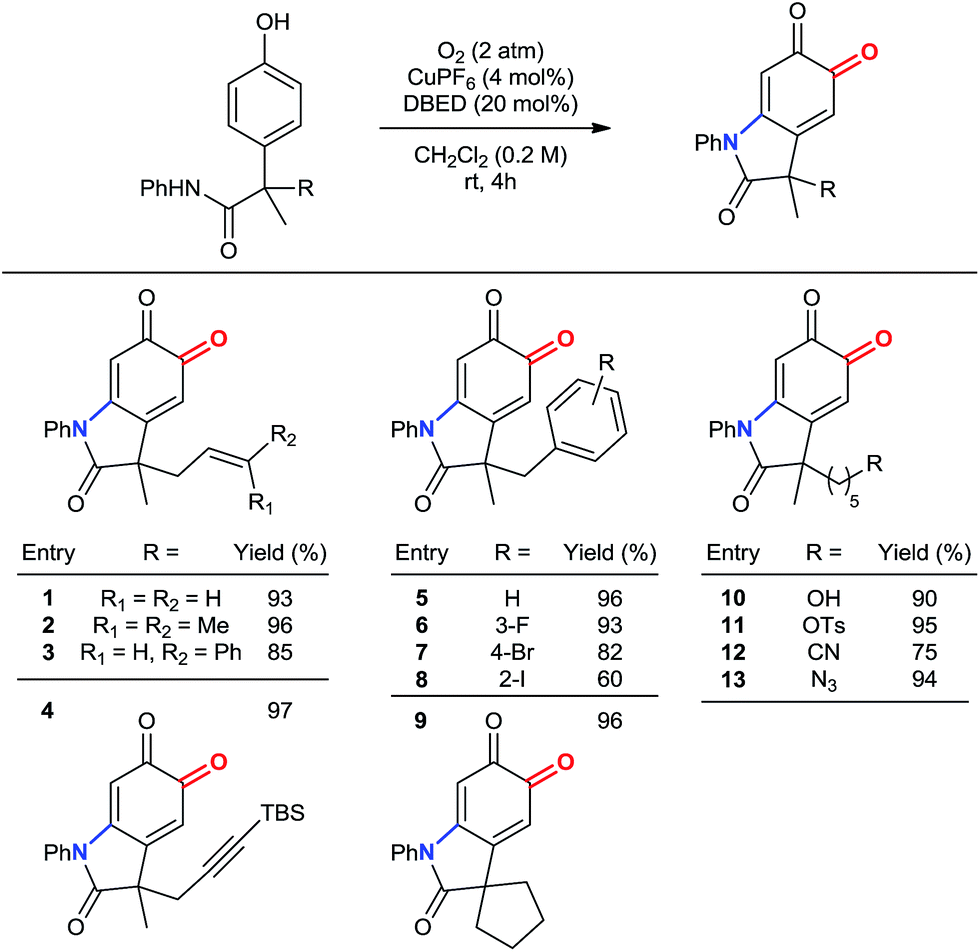
Our method remains chemoselective for ortho-oxygenation of phenols bearing a variety of benzylic substituents (Table 3). These include allyl, prenyl and cinnamyl groups (entries 1–3), as well as a silyl-protected alkyne (entry 4), demonstrating compatibility with π–bonds that provide versatile synthetic handles for additional transformations. Likewise, halogenated benzyl substituents are tolerated (entries 5–8), demonstrating chemoselectivity for phenolic oxidation in the presence of 2° benzylic hydrogen atoms. In addition to a 5-membered spirocycle (entry 9), sensitive functionalities, including a 1° alcohol, a 1° tosylate, an azide, and a nitrile are tolerated under our standard reaction conditions (entries 10–13).
Table 3 Scope of benzylic substituentsa
|
Reactions performed on 1 mmol scale and reported yields are of the purified oxindoloquinone.
|
|
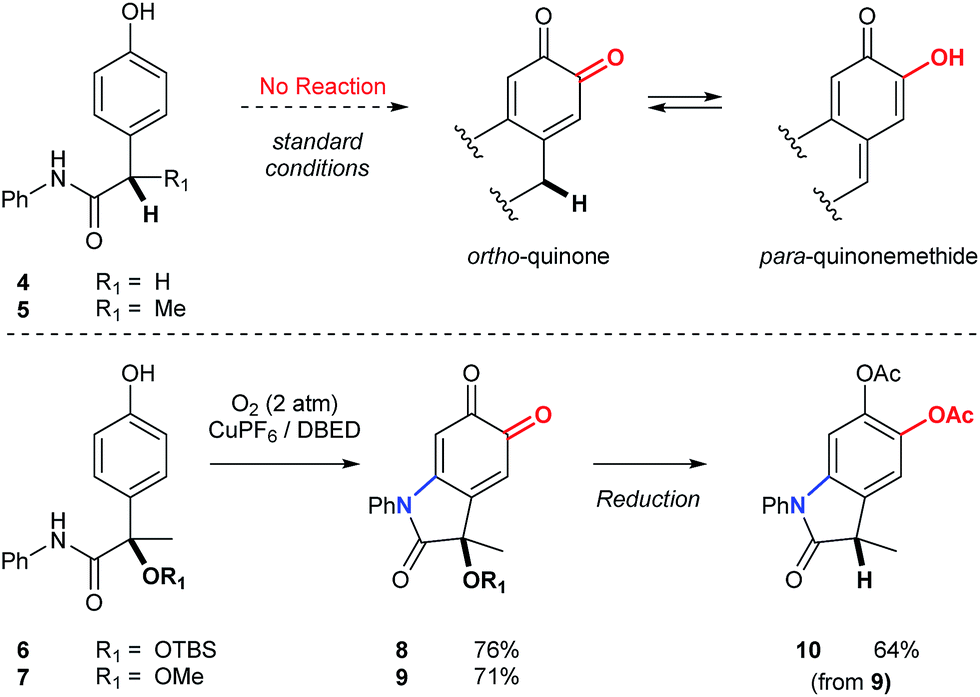
Substrates possessing hydrogen atoms α- to the amide constitute a current limitation, since tautomerization of the corresponding ortho-quinone to the para-quinone methide is problematic (Scheme 6). We have previously demonstrated that α-hydroxyketones, structurally related to the para-quinone methide, exhibit a dose-dependent inhibition of catalysis.19,40 To address this limitation in the context of an oxindole synthesis, we investigated benzylic ethers (6 and 7) as hydrogen atom surrogates, since their deoxygenation with triethylsilane occurs smoothly, following reduction and protection of oxindoloquinone 9 (Scheme 6). This provides a concise synthesis of polyfunctional oxindole 10, possessing a 3° benzylic center.
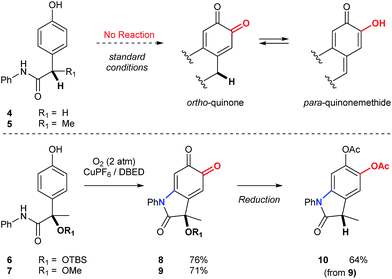 |
| | Scheme 6 Synthesis of oxindoles possessing 3° benzylic centers. Condition for the synthesis of 8 and 9: [Cu(MeCN)4](PF6) (10 mol%), DBED (50 mol%), O2 (2 atm), CH2Cl2 (0.2 M), rt, 12 h. Condition for the reduction of 9 to 10: (1) Na2S2O4(aq), then Ac2O, NEt3. (2) BF3·Et2O, Et3SiH. See ESI† for detailed experimental procedures. | |
Diversely substituted phenols.
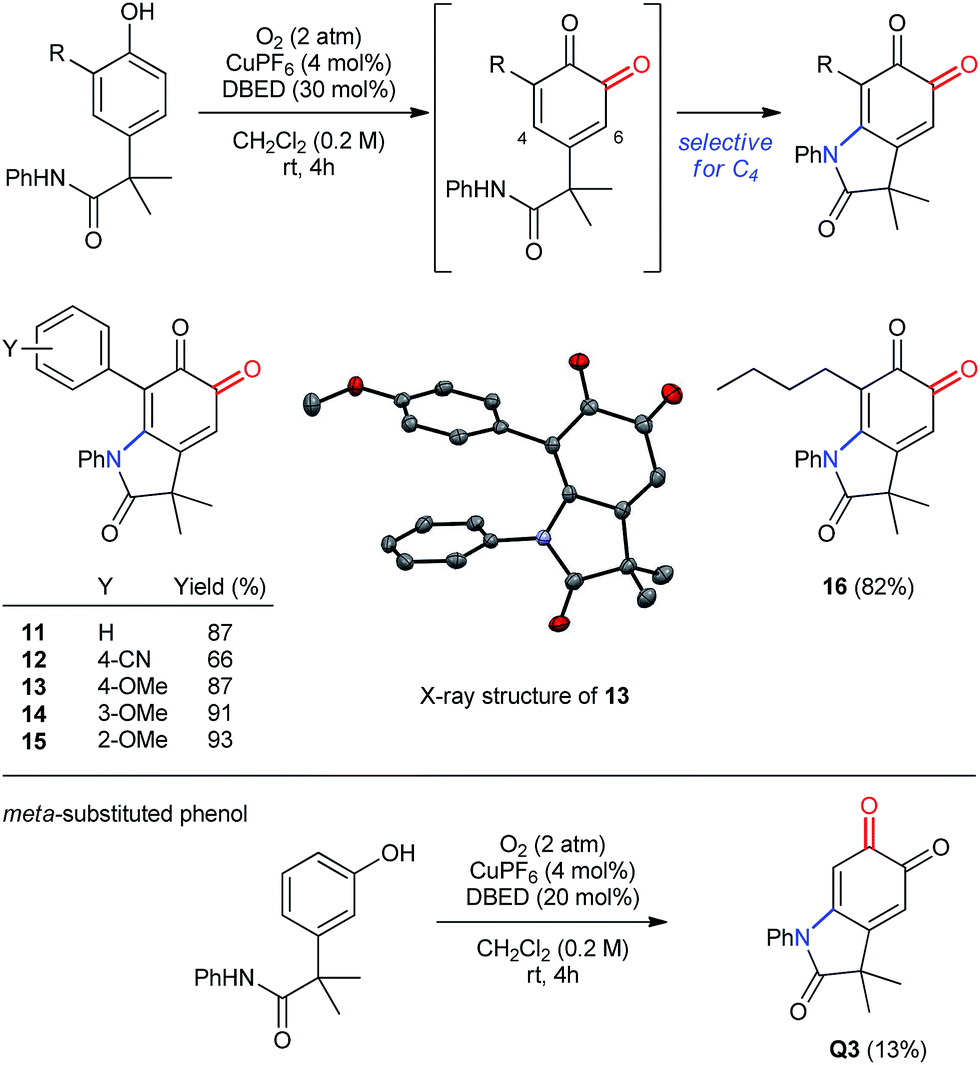
The oxygenation of more sterically encumbered 2,4-di-substituted phenols raises important questions regarding the chemo- and regioselectivity of our methodology (Scheme 7). In previous work,18a we have observed poor selectivity for ortho-oxygenation of phenols bearing tert-butyl substituents at C2. Therefore, we were pleased to observe clean oxygenation of phenols possessing less sterically demanding substituents, including aryl or alkyl groups (entries 1–6), which provide the corresponding oxindoloquinone as a single regioisomer resulting from selective 1,4-addition at C4 rather than 1,6-addition at C6. Both electron-donating and withdrawing substituents are tolerated in the para-position of the 2-aryl ring, as are methoxy groups in either ortho- or meta-positions. In addition to aryl rings, an n-butyl group is also tolerated in the ortho-position, which demonstrates compatibility for benzylic hydrogen atoms that are not acidic (entry 6). Finally, oxygenative cyclization of the meta-isomer of P1 affords an identical oxindoloquinone Q3 as is obtained from the para-isomer P1, albeit with diminished efficiency due to the formation of unidentifiable by-products.
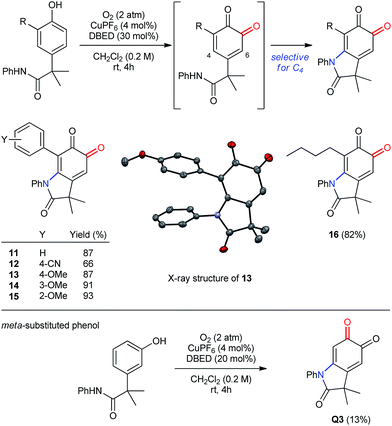 |
| | Scheme 7 Oxidation of 2,4-disubstituted and meta-substituted phenols. | |
Diversification by orthogonal C–H functionalization.
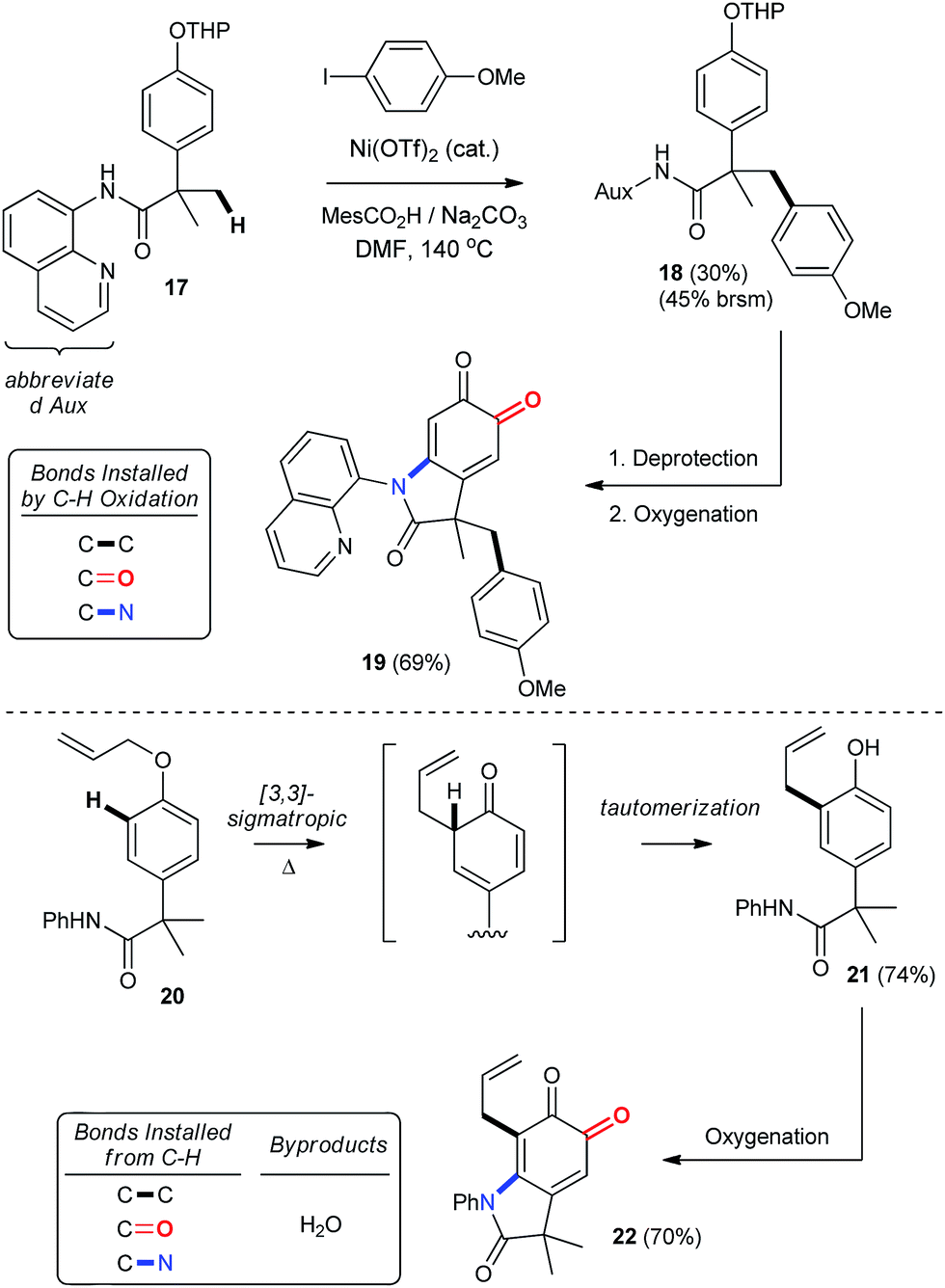
The importance of C–H functionalization to synthetic efficiency has motivated numerous strategies aimed at diversifying these relatively inert and yet omnipresent bonds.2 In this context, the 8-aminoquinoline directing group developed by Daugulis has received considerable attention for directing site-selective functionalization of both sp2 and sp3 hybridized C–H bonds.2g,39 Our chemistry offers an important complement to these previously reported methodologies, since phenolic oxygenation is not disrupted by the presence of Daugulis' group (entry 23, Table 2). This creates an opportunity to streamline substrate diversification through orthogonal C–H functionalization reactions, which we illustrate in Scheme 8 by using Chatani's Ni-catalyzed arylation of Csp3–H bonds.41 This provides THP-protected ether 18, which is readily converted into oxindoloquinone 19 following removal of the THP group and aerobic functionalization. Alternatively, a more traditional C–H functionalization by way of a 3,3-sigmatropic rearrangement can be used to synthesize 2,4-di-substituted phenol 21, which is then functionalized under our standard conditions. Thus, the syntheses of 19 and 22 highlight a unique approach to the selective functionalization of the ortho- and meta-positions of para-substituted phenols that hinges on catalytic aerobic dearomatization. This gives rise to regioselective C–C, C–O or C–N bond formation by direct C–H bond functionalization.
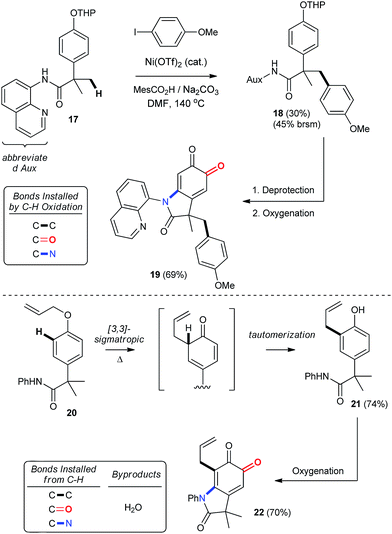 |
| | Scheme 8 Product diversification by orthogonal C–H functionalization. See ESI† for detailed experimental procedures. | |
Diversification of the oxindoloquinone.
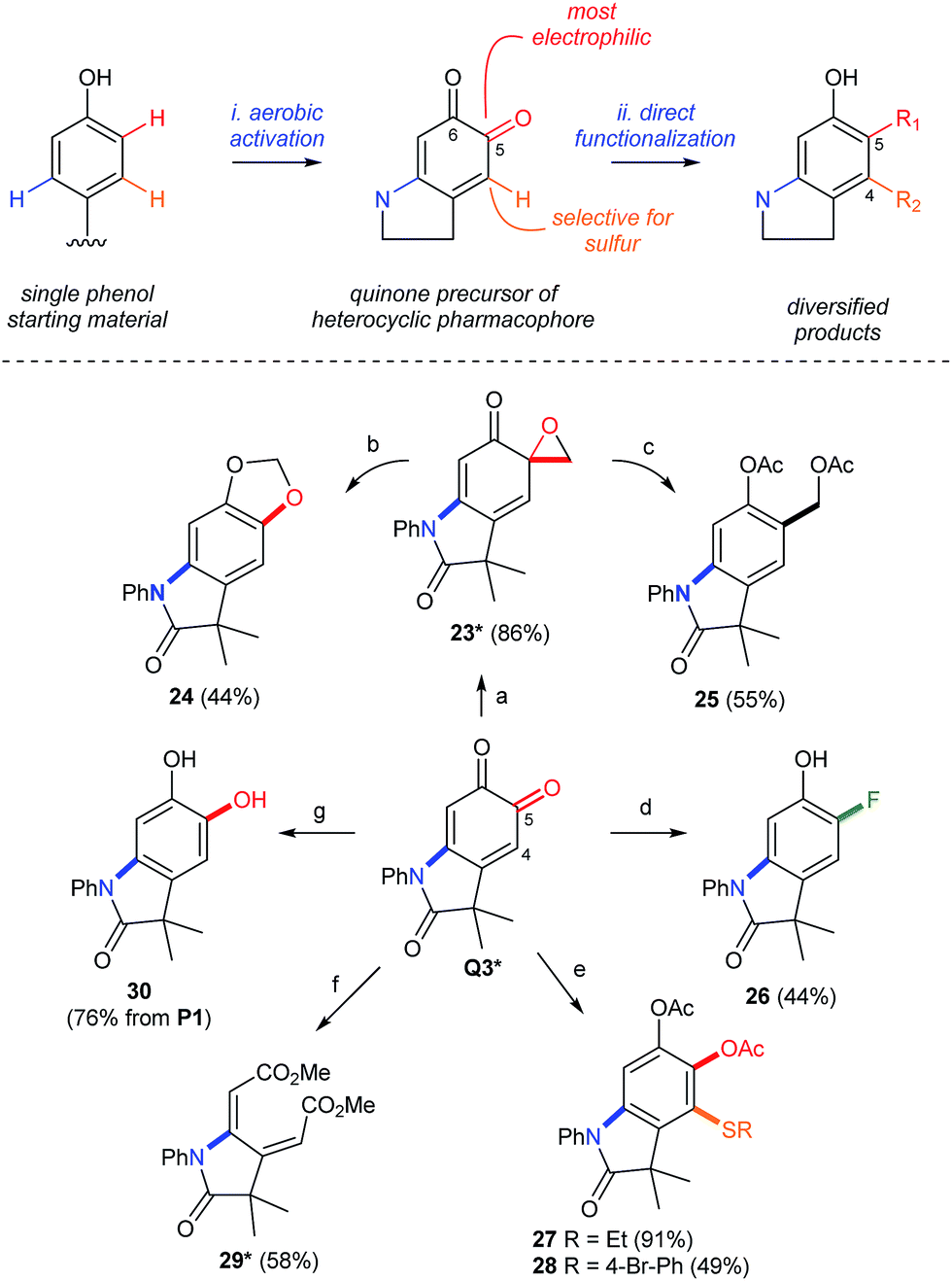
The re-aromatization of ortho-quinones provides a strong driving force to selectively diversify substituents on the corresponding oxindole. This creates a 1–3 step sequence that functionalizes the meta- and ortho-positions of the starting phenol by dual C–N/C–O, C–N/C–C, or C–N/C–F bond formation (Scheme 9). Regioselectivity is governed by the electronic differentiation of the quinone carbonyls (see top of Scheme 9). This enables selective manipulation of the more electrophilic carbonyl in Q3 by regioselective cycloproponation under the conditions of Pettus.42 The resulting α-epoxyketone 23, whose structure was confirmed by single crystal X-ray analysis (see the ESI†), is then isomerized to dioxalane 24 (ref. 43) or reduced to acetoxy methyl derivative 25 (ref. 44) by selective C–C or C–O bond cleavage, respectively. Deoxyfluorination45 is also selective for the C5 carbonyl, enabling a meta-C–N, ortho-C–F dual-functionalization of P1 over a short synthetic sequence. Differentiation of the 1,3-cyclohexadiene in Q3 is also possible by the regioselective addition of sulfur to C4, which highlights a convenient method for the formation of aromatic C–S bonds by a simple addition of the thiol at room temperature. While these examples modify the periphery of the oxindole by aromatization of Q3, cleavage of C4–C5 by lead tetraacetate46 affords the corresponding muconic ester 29 as a single geometric isomer, as determined by single crystal X-ray analysis (see the ESI†), and demonstrates that either aromatic or non-aromatic lactams, with very different three-dimensional structures, can be produced from the same starting phenol in under 2 steps. This highlights an unappreciated property of ortho-quinones, which could be attractive for applications in drug discovery where rapid diversification of a biologically active pharmacophore is required.47 We note that phenols are widely distributed in pharmaceuticals48 and natural products, making them ideal functional handles for late-stage modifications by directed C–H functionalization.
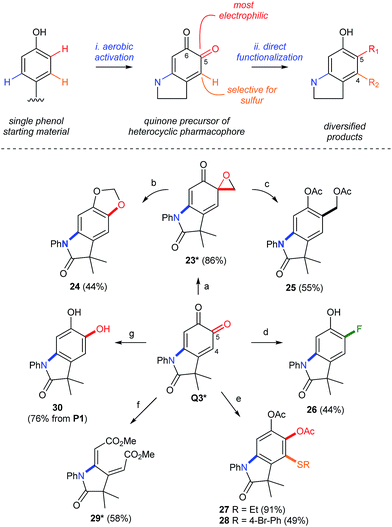 |
| | Scheme 9 Synthetic Diversification of Q3. Yields in parenthesis are based on the amount of Q3 without other notification. (a) [Cu(MeCN)4](PF6), CH2N2, THF. (b) TBSCl, NEt3, PhMe. (c) CeCl3, NaBH4, then Ac2O. (d) Deoxofluor®, CHCl3, then NaBH4, DBU, MeOH. (e) R–SH, DIPEA, CH2Cl2, then Ac2O. (f) Pb(OAc)4, PhMe/MeOH. (g) Na2S2O4 workup. *Characterized by X-ray crystallography. See the ESI† for details of the crystallographic characterization and experimental procedures. | |
Conclusion
The chemistry of melanogenesis offers a wealth of inspiration for developing low-energy and practical solutions to chemical challenges.6a This has already been appreciated in materials science, where there are countless applications of bio-inspired resins or adhesives that are synthesized by aerobic catechol or phenol polymerization reactions.13,21,49 While the value of these melanin- like materials is clear, very little is known about the precise nature of their assembly, and controlling or predictably influencing their properties remains difficult. In this article, we highlight two important considerations when approaching the chemistry of melanin. The first is the non-innocent role of transition metals following oxygenation, which influences the reactivity of ortho-quinones under our catalytic conditions. The second concerns the nucleophilic attack onto the ortho-quinone, which can trigger redox exchange. By avoiding redox exchange, our method introduces C-heteroatom bonds under exceptionally mild and selective conditions when compared to dehydrogenative coupling reactions in general.2 Finally, by generating an ortho-quinone, our method is unique amongst C–H functionalization reactions, in that the product of C–N bond formation is more reactive than the starting phenol, and is readily diversified through a series of regioselective transformations. This allows the rapid diversification of chemical space from a readily accessible starting material in a process that is solely driven by the favourable reduction of O2 to H2O.
Acknowledgements
Financial support was provided by the Natural Sciences and Engineering Council of Canada (NSERC) (Discovery Grants to J. P. L. and X. O.; Graduate Fellowship for M.S.A.; USRA Fellowship for T. Y. D.), the Fonds de Recherche du Québec-Nature et Technologies (FRQNT) (Team Grant to J. P. L and X. O.), the FRQNT Center for Green Chemistry and Catalysis at McGill University (graduate fellowship for Z. H.; summer fellowship for O. K.) and McGill University (start-up funds to J. P. L.).
Notes and references
-
(a)
L. Jiang and S. L. Buchwald, in Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH Verlag GmbH, 2008, pp. 699–760, DOI:10.1002/9783527619535.ch13;
(b)
A. M. Echavarren and D. J. Cárdenas, in Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH Verlag GmbH, 2008, pp. 1–40, DOI:10.1002/9783527619535.ch1;
(c) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS PubMed;
(d) T. Kawakami, K. Murakami and K. Itami, J. Am. Chem. Soc., 2015, 137, 2460–2463 CrossRef CAS PubMed.
-
(a) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC;
(b) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed;
(c) T.-S. Mei, L. Kou, S. Ma, K. M. Engle and J.-Q. Yu, Synthesis, 2012, 44, 1778–1791 CrossRef CAS;
(d) J. J. Song, J. T. Reeves, D. R. Fandrick, Z. Tan, N. K. Yee and C. H. Senanayake, ARKIVOC, 2010, 390–449 CAS;
(e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed;
(f) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439–2463 CrossRef CAS PubMed;
(g) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed;
(h) P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15, 5874–5883 CrossRef CAS PubMed;
(i) J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed;
(j) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed;
(k) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC.
- For a recent discussion on the difficulties of di-functionalization reactions see: H. Shi, D. J. Babinski and T. Ritter, J. Am. Chem. Soc., 2015, 137, 3775–3778 CrossRef CAS PubMed.
-
(a) B. R. Rosen, L. R. Simke, P. S. Thuy-Boun, D. D. Dixon, J.-Q. Yu and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 7317–7320 CrossRef CAS PubMed;
(b) H. Wang, G. Li, K. M. Engle, J.-Q. Yu and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6774–6777 CrossRef CAS PubMed.
-
(a) B. Trost, Science, 1991, 254, 1471–1477 CAS;
(b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS PubMed;
(c) P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505–7511 CrossRef CAS PubMed;
(d) J. B. Hendrickson, J. Am. Chem. Soc., 1971, 93, 6847–6854 CrossRef CAS;
(e) J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS;
(f) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
-
(a)
P. A. Riley, C. A. Ramsden and E. J. Land, in Melanins and Melanosomes: Biosynthesis, Structure, Physiological and Pathological Functions, ed. J. Borovanský and P. A. Riley, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, ch. 3, pp. 63–86, DOI:10.1002/9783527636150.ch3;
(b)
J. C. García-Borrón and M. C. Olivares Sánchez, in Melanins and Melanosomes, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 87–116, DOI:10.1002/9783527636150.ch4;
(c) J. D. Simon and D. N. Peles, Acc. Chem. Res., 2010, 43, 1452–1460 CrossRef CAS PubMed.
- Recent examples of C-N dehydrogenative coupling include:
(a) W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560–14561 CrossRef CAS PubMed;
(b) W. C. P. Tsang, R. H. Munday, G. Brasche, N. Zheng and S. L. Buchwald, J. Org. Chem., 2008, 73, 7603–7610 CrossRef CAS PubMed;
(c) K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931–2934 CrossRef CAS PubMed;
(d) T.-S. Mei, D. Leow, H. Xiao, B. N. Laforteza and J.-Q. Yu, Org. Lett., 2013, 15, 3058–3061 CrossRef CAS PubMed;
(e) G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944–2947 CrossRef CAS PubMed;
(f) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2011, 134, 7–10 CrossRef PubMed;
(g) T. Miura and M. Murakami, Chem. Lett., 2009, 38, 328 CrossRef CAS;
(h) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058–14059 CrossRef CAS PubMed;
(i) J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184 CrossRef CAS PubMed;
(j) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932–1934 CrossRef CAS PubMed;
(k) H. Wang, Y. Wang, C. Peng, J. Zhang and Q. Zhu, J. Am. Chem. Soc., 2010, 132, 13217–13219 CrossRef CAS PubMed;
(l) S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed;
(m) K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892–2895 CrossRef CAS PubMed;
(n) C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 1597–1600 CrossRef CAS PubMed;
(o) K. Takamatsu, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 3242–3249 CrossRef CAS PubMed;
(p) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef CAS PubMed;
(q) J. Y. Kim, S. H. Cho, J. Joseph and S. Chang, Angew. Chem., Int. Ed., 2010, 49, 9899–9903 CrossRef CAS PubMed;
(r) A. P. Antonchick, R. Samanta, K. Kulikov and J. Lategahn, Angew. Chem., Int. Ed., 2011, 50, 8605–8608 CrossRef CAS PubMed;
(s) B. Xiao, T.-J. Gong, J. Xu, Z.-J. Liu and L. Liu, J. Am. Chem. Soc., 2011, 133, 1466–1474 CrossRef CAS PubMed;
(t) E. J. Yoo, S. Ma, T.-S. Mei, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7652–7655 CrossRef CAS PubMed;
(u) C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924–18927 CrossRef CAS PubMed;
(v) R. Shrestha, P. Mukherjee, Y. Tan, Z. C. Litman and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8480–8483 CrossRef CAS PubMed;
(w) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 6043–6046 CrossRef CAS PubMed;
(x) M.-L. Louillat, A. Biafora, F. Legros and F. W. Patureau, Angew. Chem., Int. Ed., 2014, 53, 3505–3509 CrossRef CAS PubMed.
- Recent examples of C-O dehydrogenative coupling include:
(a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed;
(b) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141–1144 CrossRef CAS PubMed;
(c) X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203–12205 CrossRef CAS PubMed;
(d) C. Huang, N. Ghavtadze, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 17630–17633 CrossRef CAS PubMed;
(e) B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250–9253 CrossRef CAS PubMed;
(f) S. Bhadra, C. Matheis, D. Katayev and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 9279–9283 CrossRef CAS PubMed.
- The use of O2 as an oxygen atom transfer agent is uncommon. For examples, see:
(a) Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654–14655 CrossRef CAS PubMed;
(b) Q. Liu, P. Wu, Y. Yang, Z. Zeng, J. Liu, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 4666–4670 CrossRef CAS PubMed;
(c) Y. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827–5831 CrossRef CAS PubMed.
-
(a) E. J. Land, S. Ito, K. Wakamatsu and P. a. Riley, Pigm. Cell Res., 2003, 16, 487–493 CrossRef CAS;
(b) R. Edge, M. D'Ischia, E. J. Land, A. Napolitano, S. Navaratnam, L. Panzella, A. Pezzella, C. A. Ramsden and P. A. Riley, Pigm. Cell Res., 2006, 19, 443–450 CrossRef CAS PubMed;
(c) S. Ito and K. Wakamatsu, Photochem. Photobiol., 2008, 84, 582–592 CrossRef CAS PubMed.
-
(a)
S. Quideau, D. Deffieux and L. Pouységu, in Comprehensive Organic Synthesis II, ed. P. Knochel, Elsevier, Amsterdam, 2nd edn, 2014, pp. 656–740, DOI:10.1016/b978-0-08-097742-3.00318-9;
(b) M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077–4098 RSC;
(c) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed;
(d)
S. Yamamura, in The Chemistry of Phenols, ed. Z. Rappoport, John Wiley & Sons, Ltd, West Sussex, UK, 2003, pp. 1153–1346, DOI:10.1002/0470857277.ch17.
- M.-L. Louillat-Habermeyer, R. Jin and F. W. Patureau, Angew. Chem., Int. Ed., 2015, 54, 4102–4104 CrossRef CAS PubMed.
-
(a) A. S. Hay, Polym. Eng. Sci., 1976, 16, 1–10 CrossRef CAS PubMed;
(b) A. S. Hay, H. S. Blanchard, G. F. Endres and J. W. Eustance, J. Am. Chem. Soc., 1959, 81, 6335–6336 CrossRef CAS.
-
(a) S. Quideau, L. Pouységu, A.-V. Avellan, D. K. Whelligan and M. A. Looney, Tetrahedron Lett., 2001, 42, 7393–7396 CrossRef CAS;
(b) L. Pouységu, A.-V. Avellan and S. Quideau, J. Org. Chem., 2002, 67, 3425–3436 CrossRef PubMed;
(c) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235–2261 CrossRef PubMed;
(d) S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouységu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed;
(e) S. P. Roche and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed;
(f) S. Quideau, L. Pouységu, M. Oxoby and M. A. Looney, Tetrahedron, 2001, 57, 319–329 CrossRef CAS;
(g) S. Quideau, L. Pouysegu and D. Deffieux, Curr. Org. Chem., 2004, 8, 113–148 CrossRef CAS;
(h) G. Jacquemot, M. Menard, C. L'Homme and S. Canesi, Chem. Sci., 2013, 4, 1287–1292 RSC;
(i) H. Liang and M. A. Ciufolini, Tetrahedron, 2010, 66, 5884–5892 CrossRef CAS PubMed;
(j) H. Liang and M. A. Ciufolini, Chem.–Eur. J., 2010, 16, 13262–13270 CrossRef CAS PubMed.
- For a related examples of C-N bond formation from the corresponding catechol and a stoichiometric oxidant see:
(a) J. Clews, C. J. Cooksey, P. J. Garratt, E. J. Land, C. A. Ramsden and P. A. Riley, J. Chem. Soc., Perkin Trans. 1, 2000, 4306–4315 RSC;
(b) E. J. Land, A. Perona, C. A. Ramsden and P. A. Riley, Org. Biomol. Chem., 2009, 7, 944–950 RSC.
-
(a) P. Kovacic, R. Bennett and J. Foote, J. Org. Chem., 1961, 26, 3013–3014 CrossRef CAS;
(b) P. M. Deya, M. Dopico, A. G. Raso, J. Morey and J. M. Saa, Tetrahedron, 1987, 43, 3523–3532 CrossRef CAS;
(c) Y. Asakawa, R. Matsuda, M. Tori and M. Sono, J. Org. Chem., 1988, 53, 5453–5457 CrossRef CAS;
(d) S. Brandes, M. Bella, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 1147–1151 CrossRef CAS PubMed;
(e) S.-G. Wang, Q. Yin, C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 647–650 CAS;
(f) D. Magdziak, A. A. Rodriguez, R. W. van de Water and T. R. R. Pettus, Org. Lett., 2002, 4, 285–288 CrossRef CAS PubMed.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
-
(a) K. V. Esguerra, Y. Fall, L. Petitjean and J. P. Lumb, J. Am. Chem. Soc., 2014, 136, 7662–7668 CrossRef CAS PubMed;
(b) Y. E. Lee, T. Cao, C. Torruellas and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 6782–6785 CrossRef CAS PubMed.
- K. V. N. Esguerra, Y. Fall and J.-P. Lumb, Angew. Chem., Int. Ed., 2014, 53, 5877–5881 CrossRef CAS PubMed.
-
(a) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed;
(b) E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- S. Kobayashi and H. Higashimura, Prog. Polym. Sci., 2003, 28, 1015–1048 CrossRef CAS.
-
(a) L. M. Mirica, M. Vance, D. J. Rudd, B. Hedman, K. O. Hodgson, E. I. Solomon and T. D. P. Stack, Science, 2005, 308, 1890–1892 CrossRef CAS PubMed;
(b) B. T. Op’t Holt, M. A. Vance, L. M. Mirica, D. E. Heppner, T. D. P. Stack and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 6421–6438 CrossRef PubMed.
-
(a) M. Réglier, C. Jorand and B. Waegell, J. Chem. Soc., Chem. Commun., 1990, 1752–1755 RSC;
(b) L. Casella, M. Gullotti, R. Radaelli and P. Di Gennaro, J. Chem. Soc., Chem. Commun., 1991, 1611–1612 RSC;
(c) M. Rolff, J. Schottenheim, G. Peters and F. Tuczek, Angew. Chem., Int. Ed., 2010, 49, 6438–6442 CrossRef CAS PubMed;
(d) A. Hoffmann, C. Citek, S. Binder, A. Goos, M. Rübhausen, O. Troeppner, I. Ivanović-Burmazović, E. C. Wasinger, T. D. P. Stack and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2013, 52, 5398–5401 CrossRef CAS PubMed;
(e) J. N. Hamann and F. Tuczek, Chem. Commun., 2014, 50, 2298–2300 RSC;
(f) J. Schottenheim, C. Gernert, B. Herzigkeit, J. Krahmer and F. Tuczek, Eur. J. Inorg. Chem., 2015, 2015, 3501–3511 CrossRef CAS PubMed.
- M. S. Askari, L. A. Rodriguez-Solano, A. Proppe, B. McAllister, J. P. Lumb and X. Ottenwaelder, Dalton Trans., 2015, 44, 12094–12097 RSC.
- For a mechanistic investigation of the oxygenation of 4-tert-butyl phenol at see: M. S. Askari, K. V. N. Esguerra, J.-P. Lumb and X. Ottenwaelder, Inorg. Chem., 2015, 54, 8665–8672 CrossRef CAS PubMed.
-
(a)
K. T. Finley, in The Quinonoid Compounds 1988, John Wiley & Sons, Inc., 2010, pp. 537–717, DOI:10.1002/9780470772119.ch11;
(b) C. A. Ramsden, Adv. Heterocycl. Chem., 2010, 100, 1–51 CrossRef CAS.
- S. P. Roche and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed.
- A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed.
- For related base-mediated substitution reactions see:
(a) L. Viallon, O. Reinaud, P. Capdevielle and M. Maumy, Tetrahedron, 1996, 52, 13605–13614 CrossRef CAS;
(b) Z. Huang, O. Kwon, K. V. N. Esguerra and J.-P. Lumb, Tetrahedron, 2015, 71, 5871–5885 CrossRef CAS PubMed.
-
(a) T. R. Demmin and M. M. Rogic, J. Org. Chem., 1980, 45, 4210–4214 CrossRef CAS;
(b) K.-Q. Ling and L. M. Sayre, J. Am. Chem. Soc., 2005, 127, 4777–4784 CrossRef CAS PubMed;
(c) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. Del Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS PubMed;
(d) Z. Fu, F. Xu and H. Cai, Bioorg. Chem., 2015, 59, 31–38 CrossRef CAS PubMed;
(e) X. Guo and H. Mayr, J. Am. Chem. Soc., 2014, 136, 11499–11512 CrossRef CAS PubMed.
- M. Maumy and P. Capdevielle, J. Mol. Catal. A: Chem., 1996, 113, 159–166 CrossRef CAS.
- P. Verma, J. Weir, L. Mirica and T. D. P. Stack, Inorg. Chem., 2011, 50, 9816–9825 CrossRef CAS PubMed.
-
(a) C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 99–125 CrossRef CAS;
(b)
D. Hendrickson and C. Pierpont, in Spin Crossover in Transition Metal Compounds II, Springer Berlin Heidelberg, 2004; vol. 234, p. 63 Search PubMed.
-
(a) M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 8232–8245 CrossRef CAS;
(b) J. F. Hartwig, Inorg. Chem., 2007, 46, 1936–1947 CrossRef CAS PubMed;
(c) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852–860 CrossRef CAS.
-
(a) T.-S. Mei, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806–10807 CrossRef CAS PubMed;
(b) C. J. Vickers, T.-S. Mei and J.-Q. Yu, Org. Lett., 2010, 12, 2511–2513 CrossRef CAS PubMed.
-
(a) D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS;
(b) P. Y. S. Lam, G. Vincent, C. G. Clark, S. Deudon and P. K. Jadhav, Tetrahedron Lett., 2001, 42, 3415–3418 CrossRef CAS;
(c) D. P. Phillips, A. R. Hudson, B. Nguyen, T. L. Lau, M. H. McNeill, J. E. Dalgard, J.-H. Chen, R. J. Penuliar, T. A. Miller and L. Zhi, Tetrahedron Lett., 2006, 47, 7137–7138 CrossRef CAS PubMed;
(d) R. A. Altman, A. M. Hyde, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 9613–9620 CrossRef CAS PubMed.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS PubMed.
-
(a) J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247–1251 CrossRef CAS;
(b) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354–3357 CrossRef CAS PubMed;
(c) K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
-
(a) L. Ackermann, Acc. Chem. Res., 2013, 47, 281–295 CrossRef PubMed;
(b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed;
(c) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- Compounds with related structures are known inhibitors of tyrosinase:
(a) C. A. Ramsden and P. A. Riley, ARKIVOC, 2010, 260–274 CAS;
(b) M. R. L. Stratford, C. A. Ramsden and P. A. Riley, Bioorg. Med. Chem., 2013, 21, 1166–1173 CrossRef CAS PubMed;
(c) M. R. Loizzo, R. Tundis and F. Menichini, Compr. Rev. Food Sci. Food Saf., 2012, 11, 378–398 CrossRef CAS PubMed.
- Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS PubMed.
- L. a. Miller, M. a. Marsini and T. R. R. Pettus, Org. Lett., 2009, 11, 1955–1958 CrossRef CAS PubMed.
- J.-P. Gesson, M. Mondon and N. Pokrovska, Synlett, 1997, 12, 1395–1396 CrossRef.
- K. Hinterding, A. Knebel, P. Herrlich and H. Waldmann, Bioorg. Med. Chem., 1998, 6, 1153–1162 CrossRef CAS.
- H. Nemoto, T. Nishiyama and S. Akai, Org. Lett., 2011, 13, 2714–2717 CrossRef CAS PubMed.
- J. G. Walsh, P. J. Furlong, L. a. Byrne and D. G. Gilheany, Tetrahedron, 1999, 55, 11519–11536 CrossRef CAS.
- R. W. Huigens, K. C. Morrison, R. W. Hicklin, T. A. Flood, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195–202 CrossRef CAS PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
-
(a) Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed;
(b) H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed;
(c) Q. Liu, N. Wang, J. Caro and A. Huang, J. Am. Chem. Soc., 2013, 135, 17679–17682 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, complete characterization data. Crystallographic data for Q3, SQ3, 13, 23 and 29. CCDC 1406066-1406069 and 1421991 For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02395e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b and
Jean-Philip
Lumb
*a
*b and
Jean-Philip
Lumb
*a