
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-mediated gold-catalysed carbon(sp2)–carbon(sp) cross-coupling†
Suhong
Kim
a,
Jaime
Rojas-Martin
ab and
F. Dean
Toste
*a
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: fdtoste@berkeley.edu
bDepartamento de Química Orgánica (Módulo 01), Universidad Autónoma de Madrid, C/Francisco Tomásy Valiente 7, Cantoblanco, 28049 Madrid, Spain
First published on 10th November 2015
Abstract
A dual photoredox and gold-catalysed cross-coupling reaction of alkynyltrimethylsilanes and aryldiazonium tetrafluoroborates is described. The reaction proceeds through visible-light mediated oxidative addition of aryldiazoniums, transmetalation of alkynyltrimethylsilanes and aryl–alkynyl reductive elimination. Exclusive selectivity for silyl-substituted alkynes is observed, with no reactivity observed for terminal alkynes.
Introduction
For over a decade, homogeneous gold catalysis has been investigated because it provides access to novel modes of reactivity and enables rapid generation of complex molecular architectures.1 However, in contrast to other low-valent late transition metal catalysts, the majority of gold(I) complexes are unreactive towards the oxidative addition of aryl and vinyl halides and pseudohalides.2 Consequently, most gold-catalysed cross-coupling reactions have required sacrificial oxidants to access the +3 oxidation state.3 For example, this reactivity platform has been exploited in a gold-catalysed oxidative coupling of organosilanes and 1,2-alkene functionalizations (Scheme 1A). As an alternative, visible light-mediated oxidative addition of aryldiazoniums has emerged as a method for the generation of the requisite gold(III) intermediates via photoredox catalysis, thereby obviating the need for sacrificial oxidants.4 These gold(III) intermediates have been intercepted by a variety of nucleophiles in dual catalytic photoredox–gold reactions (Scheme 1B).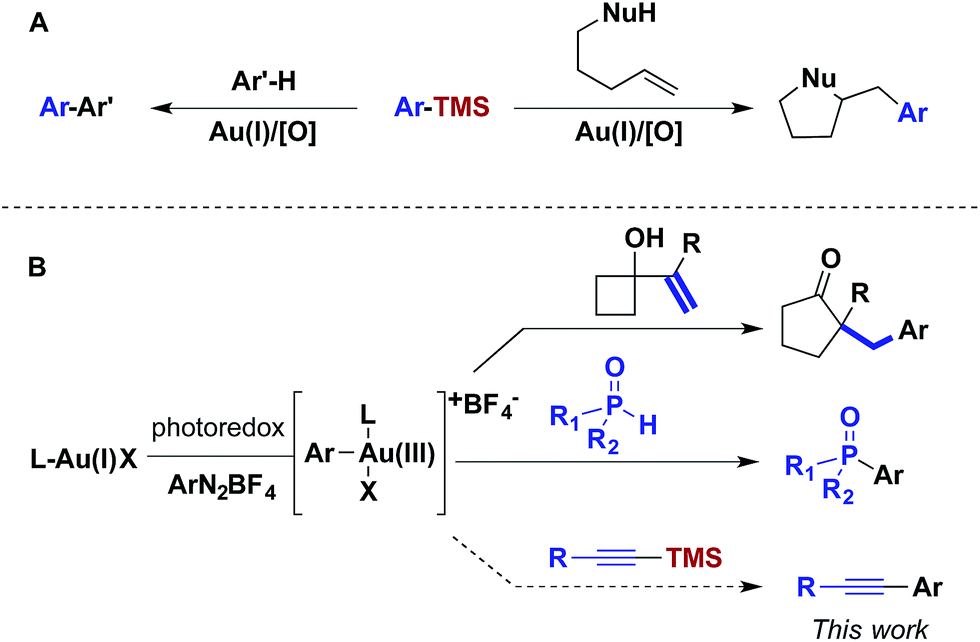 | ||
| Scheme 1 Photoredox catalyst and visible light-mediated AuI/AuIII catalysis. | ||
Although aryldiazoniums and photoredox catalysts provide an efficient method to generate gold(III) intermediates, several challenges must be addressed in order to facilitate carbon–carbon cross-coupling via these species. First, aryldiazonium salts are highly electrophilic and reactive towards many classes of nucleophilic coupling partners. Aryldiazoniums are generally decomposed under Kumada and Negishi coupling conditions and do not tolerate ligand additives and stoichiometric bases employed in the majority of Suzuki, Sonogashira and Hiyama coupling reactions.5 Second, as visible light-mediated oxidative addition of aryldiazoniums is proposed to proceed through radical intermediates, nucleophilic coupling partners that can participate in single electron transfer, such as organotin and organotrifluoroborates compounds, are problematic.6 Inspired by the oxidative coupling of organosilanes, we envisioned that the gold(III) complexes generated via photoredox catalysis might undergo transmetalation with organosilanes, generating an intermediate poised for carbon–carbon bond formation through reductive elimination.7,8
Results and discussions
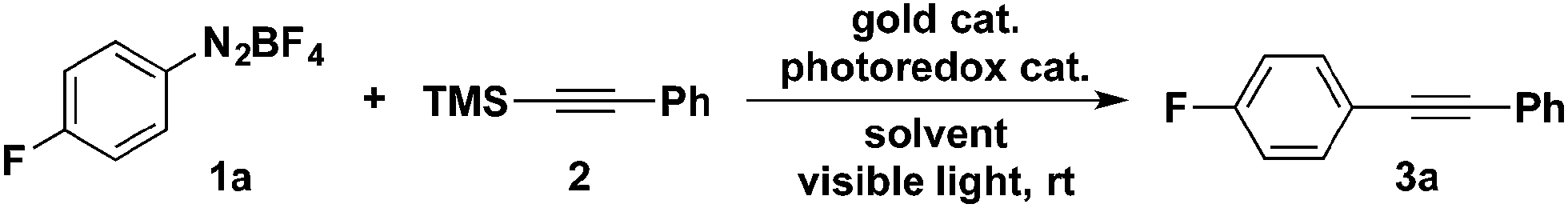
While aryldiazonium tetrafluoroborates have been shown to react with halo-, azido-, allyl- and thiotrimethylsilanes, their reactions with alkynyltrimethylsilanes has not been previously reported.9 Additionally, previous reports of organosilane transmetallation with gold lead us to hypothesize that the transmetallation of alkynyltrimethylsilanes to gold(III) intermediates might result in productive cross-coupling. On the basis of this hypothesis, we examined the combined gold/photoredox coupling of 1 equivalent of aryldiazonium salt 1a and 1 equivalent of alkynyltrimethylsilane 2.Evaluation of reaction conditions showed that the combination of Ar3PAuCl and Ru(bpy)3(PF6)2, in acetonitrile gave the highest yield of aryl alkyne 3a (Table 1). Changing either the solvent or the photoredox catalyst proved detrimental to the yield of the desired product. With respect to the ligands on the gold catalyst, both electronic and steric factors impacted the efficiency of the cross-coupling. For example, electron-deficient triarylphosphine ligands resulted in significantly lower yield of 3a (Table 1, entry 10). The sterics of the ligand showed an even more dramatic effect on reaction yield. The reaction conducted with (p-tol)3PAuCl provided 72% yield of the desired product, while the much more hindered (o-tol)3PAuCl-catalysed reaction only provided 17% yield of 3a (Table 1, entry 11 and 12). The yield of product was lower when tricyclohexylphosphinegold(I) chloride was used as catalyst instead of triphenylphoshinegold(I) chloride (Table 1, entry 13). Gold(I) complexes of dialkylbiarylphosphine were also examined, but all showed less than 5% yield due to the combined effect of dialkyl groups and the steric effect of biaryl groups. Finally, no coupling product was observed in the absence of the ruthenium catalyst (entry 14) or when the reaction was conducted in the dark (entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | Gold cat. | Photoredox cat. | Solvent | Yielda |
| Reaction conditions: 4-fluorobenzenediazonium tetrafluoroborate 1a (0.2 mmol, 1 equiv.), 1-trimethylsilyl-2-phenylacetylene 2 (0.2 mmol, 1 equiv.), gold catalyst (5 mol%), photoredox catalyst (1 mol%), solvent (2 mL), N2 atmosphere, visible light, rt for 6 h.a GC yields.b The reaction was run in the dark. bpy = 2,2′-bipyridine, ppy = 2-phenylpyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. | ||||
| 1 | Ph3PAuCl | Ru(bpy)3(PF6)2 | CH3CN | 70 |
| 2 | Ph3PAuCl | Ru(bpy)3(PF6)2 | Acetone | 39 |
| 3 | Ph3PAuCl | Ru(bpy)3(PF6)2 | CH3NO2 | 15 |
| 4 | Ph3PAuCl | Ru(bpy)3(PF6)2 | DMF | 10 |
| 5 | Ph3PAuCl | Ru(bpy)3(PF6)2 | MeOH | 18 |
| 6 | Ph3PAuCl | Ru(bpy)3(PF6)2 | EtOH | 4 |
| 7 | Ph3PAuCl | Ir(ppy)3 | CH3CN | 21 |
| 8 | Ph3PAuCl | Ir(ppy)2(dtbbpy)PF6 | CH3CN | 45 |
| 9 | (p-MeOPh)3PAuCl | Ru(bpy)3(PF6)2 | CH3CN | 73 |
| 10 | (p-CF3Ph)3PAuCl | Ru(bpy)3(PF6)2 | CH3CN | 43 |
| 11 | (p-tol)3PAuCl | Ru(bpy)3(PF6)2 | CH3CN | 72 |
| 12 | (o-tol)3PAuCl | Ru(bpy)3(PF6)2 | CH3CN | 17 |
| 13 | Cy3PAuCl | Ru(bpy)3(PF6)2 | CH3CN | 48 |
| 14 | Ph3PAuCl | — | CH3CN | 0 |
| 15b | Ph3PAuCl | Ru(bpy)3(PF6)2 | CH3CN | 0 |
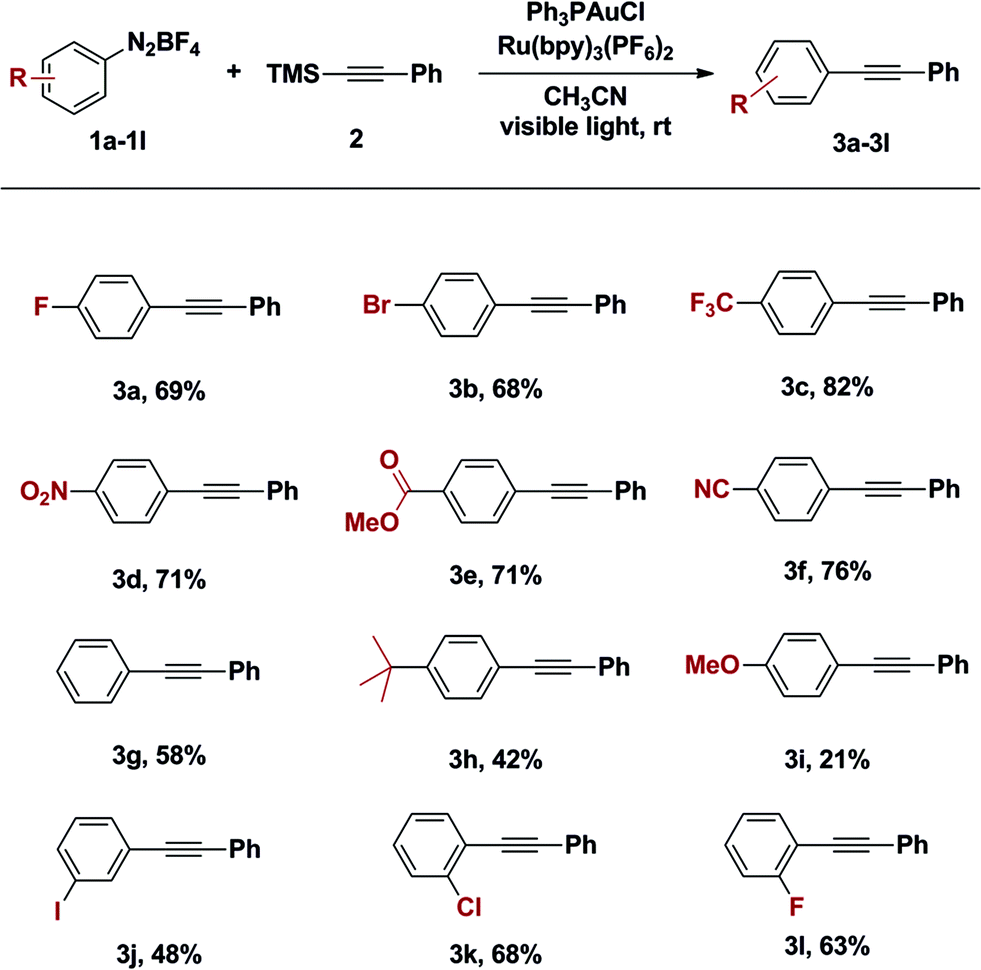
With an optimized catalyst system in hand, the scope with respect to the aryldiazonium coupling partner was examined (Table 2). The yields of alkynylation reactions of electron-poor aryldiazonium salts were generally higher than those of reactions with electron-rich aryldiazonium coupling partners. It should be noted that the intrinsic instability of ortho-substituted aryldiazonium salts limited their use in this transformation (Table 2, entry 3k and 3l). It is also noteworthy that all halogen substitutions on the aryldiazonium coupling partner were preserved during the coupling reaction. Bromo- and iodoaryldiazonium salts were readily coupled and leaving halides intact for use in further reactions (Table 2, 3b and 3j); this chemoselectivity is challenging under typical palladium-catalysed Sonogashira coupling reactions.10
| Reaction conditions: 1a–l (0.24 mmol, 1.2 equiv.), 2 (0.2 mmol, 1.0 equiv.), Ph3PAuCl (10 mol%), Ru(bpy)3(PF6)2 (2 mol%), acetonitrile (2 mL), N2 atmosphere, visible light, rt for 3 h. Isolated yields. |
|---|
|
The difference in reactivity of electron-rich and -poor aryldiazoniums may be the result of competing reaction pathways. Oxidative addition is believed to proceed through single electron reduction of aryldiazoniums and this process is preferred with electron-poor aryldiazonium salts.4 On the other hand, the generation of aryl cations, through loss of dinitrogen, is facile with electron-rich aryldiazoniums. Correspondingly, we observed trace amount of desired product when benzenediazonium tetrafluoroborate was heated at 60 °C for 9 hours in the presence of 1-trimethylsilyl-2-(4-methoxy)phenylacetylene; however the same reactivity was not observed with 4-fluorophenyldiazonium salts on the same conditions.
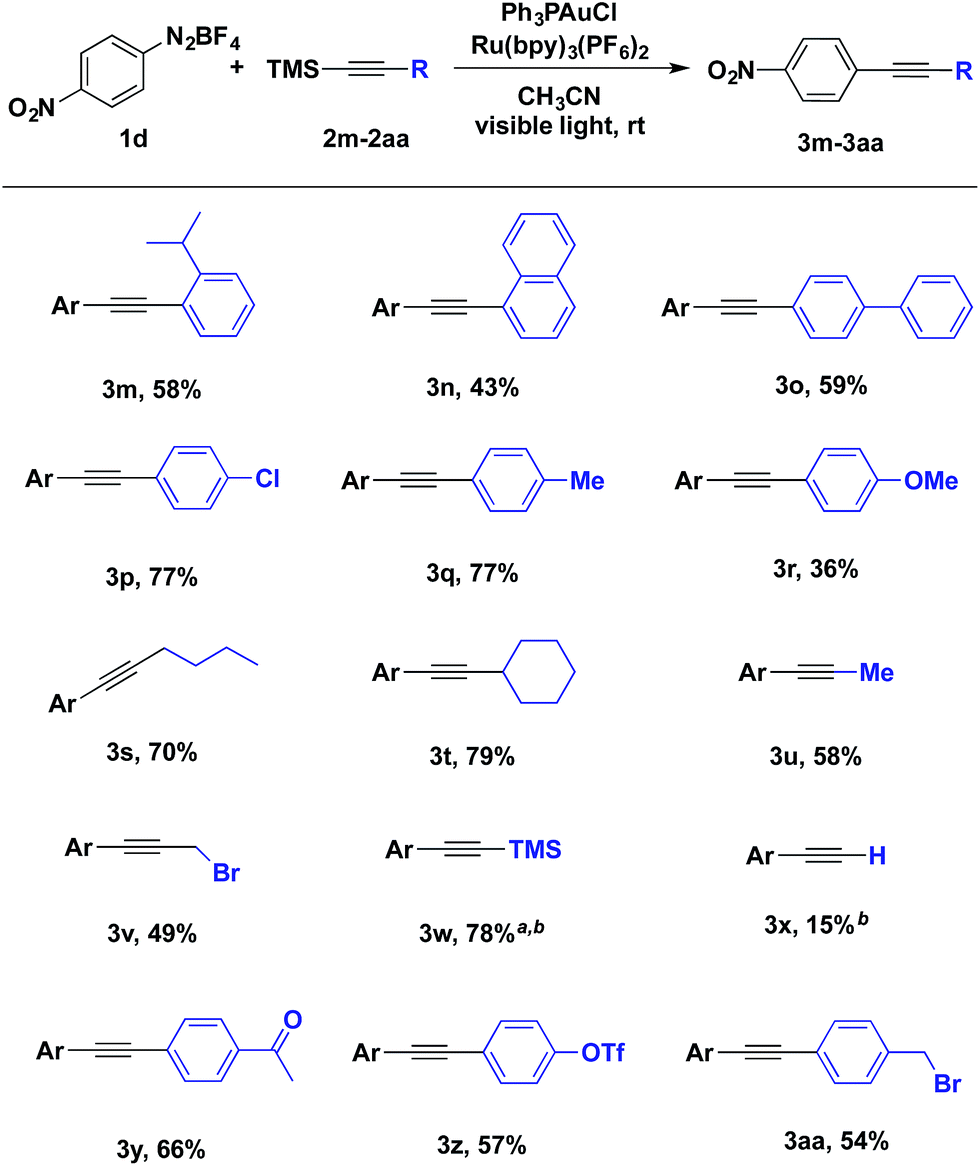
The scope of alkynyltrimethylsilanes was also investigated (Table 3). Both aryl- and alkylethynyltrimethylsilanes were coupled with modest to high product yields. o-Isopropylphenyl, biphenylyl and naphthylalkynyltrimethylsilanes participated in the gold-catalysed coupling (Table 3, 3m, 3n and 3o). Potentially sensitive benzylic and propargylic C–H and C–X bonds were well tolerated under the reaction conditions (Table 3, 3q, 3aa and 3s–v). Additionally, alkynyltrimethylsilane 3w was prepared in 78% yield from the coupling of aryldiazonium salt 1d and bis(trimethylsilyl)acetylene, providing a compliment to using trimethylsilylacetylene in a traditional Sonogashira coupling (Table 3, 3w).
| Reaction conditions: 1d (0.24 mmol, 1.2 equiv.), 2m–aa (0.2 mmol, 1.0 equiv.), Ph3PAuCl (10 mol%), Ru(bpy)3(PF6)2 (2 mol%), acetonitrile (2 mL), N2 atmosphere, visible light, rt for 3 h. Isolated yields. Ar = p-nitrophenyl.a 5 equiv. of bis(trimethylsilyl)acetylene was used with 1 equiv. of 1d.b GC yields. |
|---|
|
Another interesting feature of this reaction is the effect of the silyl group on yield (Table 4). When trimethylsilylacetylene was coupled with 1d to generate terminal alkyne 3x, the yield was 15% with none of the corresponding silylacetylene 3w (Table 3). This observation suggests that, under the current base free reaction conditions, the silyl group plays a critical role in the coupling. To investigate this role, a set of reactions was performed by varying counteranions of aryldiazonium salts and silyl group identities of alkynylsilanes. The yield of coupling product was diminished as the steric hindrance of silyl group was increased (Table 4, entry 1 and entries 4–7). While both tetrafluoroborate and tosylate diazonium salts proved efficient coupling partners, the yield was diminished when hexafluorophosphate salts were used.11 Moreover, when the corresponding terminal alkyne was used instead of alkynyltrimethylsilanes, only 3% of the coupling product was observed (Table 4, entry 8).12 Taken together, these results suggests that transmetallation from the organosilane is critical for high efficiency of the current coupling reaction.
|
|
|||
|---|---|---|---|
| Entry | Counteranion (X) | Silyl group (Y) | Yielda |
| Reaction conditions: aryldiazonium salt (0.2 mmol, 1 equiv.), phenylethynylsilane (0.2 mmol, 1 equiv.), Ph3PAuCl (5 mol%), Ru(bpy)3(PF6)2 (1 mol%), acetonitrile (2 mL), N2 atmosphere, visible light, rt for 6 h.a GC yields. | |||
| 1 | BF4− | TMS | 70 |
| 2 | TsO− | TMS | 68 |
| 3 | PF6− | TMS | 24 |
| 4 | BF4− | TES | 61 |
| 5 | BF4− | TBDMS | 10 |
| 6 | BF4− | TIPS | 10 |
| 7 | BF4− | TBDPS | 1 |
| 8 | BF 4 − | H | 3 |

Other organotrimethylsilanes were tested under the optimized reaction conditions. While the reactions of aryl and vinylsilanes were complicated by competing reactions,13,14 the gold/photoredox-catalysed coupling of allenyltrimethylsilane 4 provided propargylic compound 5 as the sole coupling product (Scheme 2). The product from aryl–allenyl reductive elimination and the other aryl–propargylic coupling isomer were prepared independently (Scheme 2, 6 and 7). We examined whether 6 was an intermediate that underwent isomerization to 5 facilitated by visible light and the photoredox catalyst. However, no isomerization was observed of 6 to either 5 or 7 under the reaction conditions. Therefore, the formation of 5 is best rationalized by transmetalation of 4 to the gold(III) intermediate, followed by 1,3-migration and aryl–propargyl reductive elimination.
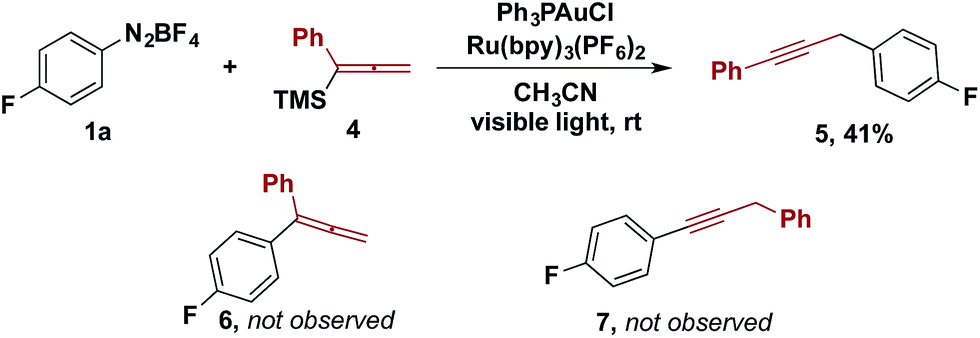 | ||
| Scheme 2 Reaction with allenyltrimethylsilanes. | ||
Conclusions
In summary, we have demonstrated that trimethylsilylalkynes and aryldiazonium tetrafluoroborates can be coupled via dual photoredox and gold catalysis strategy. The reaction proceeds under mild conditions and shows excellent functional group tolerance, including aryl halides that may be reactive under traditional coupling conditions. This process compliments the Sonogashira coupling, especially when aryl–alkynyl coupling is required under non-basic conditions and may present an advantage when TMS-alkynes are available rather than corresponding terminal alkynes.Acknowledgements
We are grateful for financial support from the NIHGMS (RO1 GM073932). Jaime Rojas-Martin thanks MINECO for the EEBB-FPI fellowship. We thank Mark D. Levin, William J. Wolf and Matthew S. Winston for helpful discussions.Notes and references
- For recent reviews on gold catalysis: (a) A. S. K. Hashimi, Acc. Chem. Res., 2014, 47, 864 CrossRef PubMed; (b) C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16 RSC; (c) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS; (d) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC; (e) L. P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129 RSC; (f) N. D. Shapiro and F. D. Toste, Synlett, 2010, 675 CAS.
- (a) T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006 CrossRef CAS PubMed. For recent breakthroughs, (b) M. D. Levin and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 6211 CrossRef CAS PubMed; (c) M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777 CrossRef CAS PubMed; (d) J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778 CrossRef CAS PubMed; (e) M. Joost, A. Zeineddine, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed.
- For early publications on gold-catalysed oxidative coupling, (a) A. Kar, N. Mangu, H. M. Kaiser, M. Beller and M. K. Tse, Chem. Commun., 2008, 386 RSC; (b) G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112 CrossRef CAS PubMed; (c) A. D. Melhado, W. E. Brenzovich, A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885 CrossRef CAS PubMed; (d) W. E. Brenzovich, D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 5519 CrossRef PubMed; (e) W. E. Brenzovich, J. F. Brazeau and F. D. Toste, Org. Lett., 2010, 12, 4728 CrossRef CAS PubMed; (f) G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474 CrossRef CAS PubMed; (g) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS PubMed; (h) J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (i) J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 7304 CrossRef CAS PubMed; (j) M. N. Hopkinson, A. Tessier, A. Salisbury, G. T. Giuffredi, L. E. Combettes, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2010, 16, 4739 CrossRef CAS PubMed; (k) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Org. Lett., 2010, 12, 4724 CrossRef CAS PubMed; (l) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed.
- For visible-light mediated oxidative addtion of aryldiazoniums on gold catalysis, (a) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (b) M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794 CrossRef CAS; (c) X. Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed; (d) Y. He, H. Wu and F. D. Toste, Chem. Sci., 2015, 6, 1194 RSC.
- A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed.
- For single-electron transmetalation of organotrifluoroborates (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (b) D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS PubMed.
- W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2013, 6, 159 CrossRef PubMed.
- For cross-coupling reactions of alkynyltrimethylsilanes, (a) S. A. Lermontov, I. M. Rakov, N. S. Zefirov and P. J. Stang, Tetrahedron Lett., 1996, 37, 4051 CrossRef CAS; (b) G. W. Kabalka, L. Wang and R. M. Pagni, Tetrahedron, 2001, 57, 8017 CrossRef CAS; (c) S. Kang, H. Ryu and Y. Hong, J. Chem. Soc., Perkin Trans. 1, 2007, 736 Search PubMed; (d) C. Yang and S. P. Nolan, Organometallics, 2002, 21, 1020 CrossRef CAS; (e) L. K. Hwang, Y. Na, J. Lee, Y. Do and S. Chang, Angew. Chem., Int. Ed., 2005, 44, 6166 CrossRef CAS PubMed; (f) W. J. Sommer and M. Weck, Adv. Synth. Catal., 2006, 348, 2101 CrossRef CAS; (g) N. Sakai, R. Komatsu, N. Uchida, R. Ikeda and T. Konakahara, Org. Lett., 2010, 12, 1300 CrossRef CAS PubMed; (h) Y. Nishihara, S. Noyori, T. Okamoto, M. Suetsugu and M. Iwasaki, Chem. Lett., 2011, 40, 972 CrossRef CAS; (i) Y. Nishihara, D. Ogawa, S. Noyori and M. Iwasaki, Chem. Lett., 2012, 41, 1503 CrossRef CAS; (j) L. Peng, F. Xu, Y. Suzuma, A. Orita and J. Otera, J. Org. Chem., 2013, 78, 12802 CrossRef CAS PubMed.
- For reactions of aryldiazonium tetrafluoroborate with halo-, azido-, allyl- and thiotrimethylsilanes, (a) T. Keumi, T. Umeda, Y. Inoue and H. Kitajima, Bull. Chem. Soc. Jpn., 1989, 62, 89 CrossRef CAS; (b) H. Mayr and K. Grimm, J. Org. Chem., 1992, 57, 1057 CrossRef CAS; (c) G. K. S. Prakash, D. Hoole, D. S. Ha, J. Wilkinson and G. A. Olah, ARKIVOC, 2002, 13, 50 Search PubMed.
- For Sonogashira cross-coupling of aryldiazonium salts, (a) S. Darses, G. Michaud and J. Genet, Eur. J. Org. Chem., 1999, 1875 CrossRef CAS; (b) G. Fabrizi, A. Goggiamani, A. Sferrazza and S. Cacchi, Angew. Chem., Int. Ed., 2010, 49, 4067 CrossRef CAS PubMed; (c) B. Panda and T. K. Sarkar, Chem. Commun., 2010, 46, 3131 RSC; (d) X. Wu, H. Neumann and M. Beller, Chem. Commun., 2011, 47, 7959 RSC; (e) This challenge was overcome by employing o-benzenedisulfonimide as a counteranion. M. Barbero, S. Cadamuro and S. Dughera, Eur. J. Org. Chem., 2014, 598 CrossRef CAS PubMed; (f) While preparing a manuscript, gold-catalysed cross-coupling of aryldiazonium salts with terminal alkynes and boronic acids was published. R. Cai, M. Lu, E. Y. Aguilera, Y. Xi, N. G. Akhmedov, J. L. Petersen, H. Chen and X. Shi, Angew. Chem., Int. Ed., 2015, 54, 8772 Search PubMed.
- After the completion of coupling reaction with tetrafluoroborate salts, stoichiometric amounts of trimethylsilyl fluoride were observed by 1H NMR.
- To examine the robustness of this reaction towards terminal alkynes, 1 equiv. of phenylacetylene and 1 equiv. of phenylethynyltrimethylsilane were reacted with 1 equiv. 1a surprisingly, less than 5% of 3a was detected.
- When (Z)-styryltrimethylsilane was used the corresponding Z-product was obtained from gold-catalysed cross coupling and E-product was obtained from uncatalysed addition of the aryldiazonium to the vinylsilane (see: A. Benkeser, E. W. Bennet and R. A. Hickner, J. Am. Chem. Soc., 1957, 79, 6253 CrossRef .) After consumption of the starting material, photoredox promoted isomerization of E-stilbene to Z-stilbene occurred.
- In the case aryltrimethylsilanes, further optimization is required due to the formation of significant amounts of competing homo-coupling reactions.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03025k |
| This journal is © The Royal Society of Chemistry 2016 |