
Gas–solid conversion of lignin to carboxylic acids
Samira
Lotfi
*,
Daria C.
Boffito
 and
Gregory S.
Patience
*
and
Gregory S.
Patience
*
Department of Chemical Engineering, École Polytechnique de Montréal, P.O. Box 6079, C.P. Centre-ville, Montréal H3C 3A7 (Canada). E-mail: samira.lotfi@gmail.com; gregory-s.patience@polymtl.ca.; Fax: +1 514 340 4059; Tel: +1 514 340 4711
First published on 11th May 2016
Abstract
Lignin represents 15% to 40% of the dry weight of lignocellulosic biomass but remains under exploited as a sustainable feedstock for chemical and fuels even though it is the only bio-polymer with aromatic units. Technology that selectively converts lignin to value added specialities would improve the economics of the burgeoning bio-refinery industry. Here, we introduce a two stage gas-phase catalytic process that produces carboxylic acids and aromatics from lignin while minimizing coke and char and maintaining catalyst activity. In the first step, a mixture of 50% water vapour in air crack and partially oxidize this complex macromolecule (<550 °C). The effluent gas contacts heterogeneous mixed-metal oxides or metal catalysts in the second step. The product profile from the second step includes aromatic compounds but mostly C4 carboxylic acids such as maleic acid and butyric acid. Vanadium catalysts cleave lignin bonds, open aromatic rings and oxidize lignin to carboxylic acids, especially maleic acid. WO3/TiO2 mostly gave butyric acid. Basic catalysts produced more aromatic compounds. The maximum amount of coke was 5% of the total carbon in lignin.
1 Introduction
Bio-refineries that convert lignocellulosic biomass into fuels and chemicals represent a paradigm shift versus the pulp and paper industry that dominates the market. Lignocellulosics contains cellulose, hemicellulose and lignin macromolecules.1 Cellulose and hemicellulose are raw materials for bioethanol, bio-butane and other chemicals.2–4 The pulp and paper industry combusts as much as 40% of their lignin. Only 2% is converted to value-added chemicals3 and the rest is waste—50 million tons per year.2,5 According to 2007 U.S. Energy Security and Independence Act, 79 million m3 of biofuels from biomass generates 62 million tons of lignin as a residue.3Methoxylated phenylpropane units linked by C–C and ether bonds constitute the skeleton of lignin as a bio-polymer.2 Lignin accounts for 15% to 40% of lignocellulose, but the heterogeneity of its structure converts it to an uncommon feedstock for chemicals or even liquid fuels.6,7 Moreover, chemicals processes require a homogeneous feedstock to minimize multiple purification trains that engender costs and complexity for biorefiners.2,6
Pyrolysis, gasification, hydrogenation, oxidation, aqueous reforming, hydrolysis and enzymatic processes convert lignin to aromatic compounds.1,2,8–10 The products include low molecular weight phenolic compounds, carboxylic acids and quinones. Depolymerization of lignin leads to fuels rather than chemicals and includes a complex mixture of molecules. Oxidative cleavage of lignin mainly yields polyfunctional aromatic compounds including aromatic aldehydes and carboxylic acids.11
The market for aromatic acids and aldehydes is smaller, while the market for ring-opened, high purity aliphatic acids is much larger and includes pharmaceuticals, food, petrochemicals and polymers.3,5,6 One of the challenges to oxidatively cleave lignin is to limit the further oxidation of the liquid products of interest to gas.12
Heterogeneous catalysis plays a key role in the manufacture of polymers, agricultural and pharmaceuticals, especially in selective oxidation and hydrogenation.13–16 Catalysts improve the selectivity to target molecules and reduce the activation energy so that the process operates at lower temperature. Developing a selective catalytic process to convert lignin to chemicals and fuel represents a paradigm shift with respect to current practice in the pulp and paper industry17 and could ensure the economic viability of biorefineries. Several catalytic systems have been designed for lignin model compounds that are ineffective for real biomass substrates. For example, Pd/C depolymerises up to 98% of dimeric lignin model compounds, but not real lignin.18 Lignin forms coke and char and other compounds that deactivate catalysts.2,19
Catalysts that oxidatively cleave lignin include organometalics, metal-free organic catalysts, acid and base catalysts, metal salt catalysts, photo- and electro catalysts.11 H2O2 and chalcopyrite-CuFeS2 in an acetic acid buffer oxidized lignin to malonic, succinic, malic and maleic acid at 333 K. Ma et al. propose a molecular pathway where HO+ initiates the ring hydroxylation to depolymerize lignin. The reaction also produced armotic compounds—benzoic acid, vanillin and vanillic acid. Further oxidation yields p- and o-quinone, which can undergo ring opening to give C4 acids such as maleic, fumaric and muconic acid. Hydrolyzing these acids gave stable endpoint acids (malonic, succinic, malic and trace of maleic acid).17 Vardon et al.6 produced adipic acid from lignin model compounds. First, a fed-batch biological cultivation process converted the model compound to cic–cis muconate acid. Then PdC catalyst hydrogenated the recovered acid to adipic acid.
Olcese et al.20 improved the quality of lignin pyrolysis bio-oil by hydrotreating the vapours over iron–silica (Fe/SiO2) and iron–activated carbon (Fe/AC) catalysts in a fixed bed at 400 °C and 1 bar. The products were benzene, toluene, xylenes, phenol, cresols, and alkyl phenols. Coke plugged the micropores of Fe/AC but not the mesopores of the Fe/SiO2.20
Fan et al.2 converted lignin to ethylbenzene in a two-step process. In the first step, zeolites depolymerized lignin to aromatics (mainly benzene). In the second step, HZSM-5 (with ethanol) selectively alkylated the aromatic compounds to ethylbenzene. Large pores reduced the benzene selectivity and yield of benzene, toluene, and xylenes increased with catalyst acidity. The catalyst deactivated after 3 uses: the selectivity to benzene dropped from 23% to 7% but a 4 h oxygen treatment restored the original catalytic activity.2 Alternating reaction/regeneration cycles is a successful strategy to maintain catalyst activity over time and reduce deactivation.21
Lignin pyrolysis yields phenolic compounds that are char precursors.22 Here, we introduce a new reactor configuration that includes thermo-oxidative cracking of lignin in the gas phase, followed by catalytic oxidation in a gas–solids catalytic reactor. The products are fine chemicals, particularly carboxylic acids. To reduce coking and the regeneration cycles, we developed four micro-reactor configurations to activate lignin involving air, steam and heterogeneous catalysts. Based on screening tests, passing the effluent gases after a thermo-oxidative steam cracking step produced the most organic acids and phenolic compounds.
2 Experimental
2.1 Materials and catalyst preparation
We identified the structural signature of the softwood kraft lignin by 13C NMR, which identifies most of the carbon groups making up lignin25 while other methods can also quantify the functional groups.26 Most of the contributions identified belong to the β-O-4 type linkage that characterize up to 50% of softwood lignin. The spectral region from 185 to 164 ppm represents the C(O)O–(H,R) groups of carboxylic acids and esters.27 The sharp peak at δ = 147 ppm belongs to C-4 in etherified guaiacyl (G) units and C-3 in non etherified G units (β-O-4 type). Resonances at ∼115 and ∼127 ppm belong to carbons 3/5 (m) and 2/6 (o) and signals between 157 and 152 ppm to the quaternary carbons in p-position.28 Signal at ∼87 and ∼83 ppm belong to G units and represent C-α in the β-5 linkage and C-β in the β-O-4 linkage, respectively. Signals at 71.8 and 71.2 ppm belong to G units and correspond to C-α in β-O-4 linkages and C-γ in β–β linkages. The sharp peak at δ = 55.6 ppm identifies C in Ar–OCH3.29 Peaks from 53 to 49 ppm represent CH2O of esters.25 Signals from 49 to 5 ppm belong to CH, CH2, CH3 aliphatic unbound with an oxygen atom. (Fig. 1).
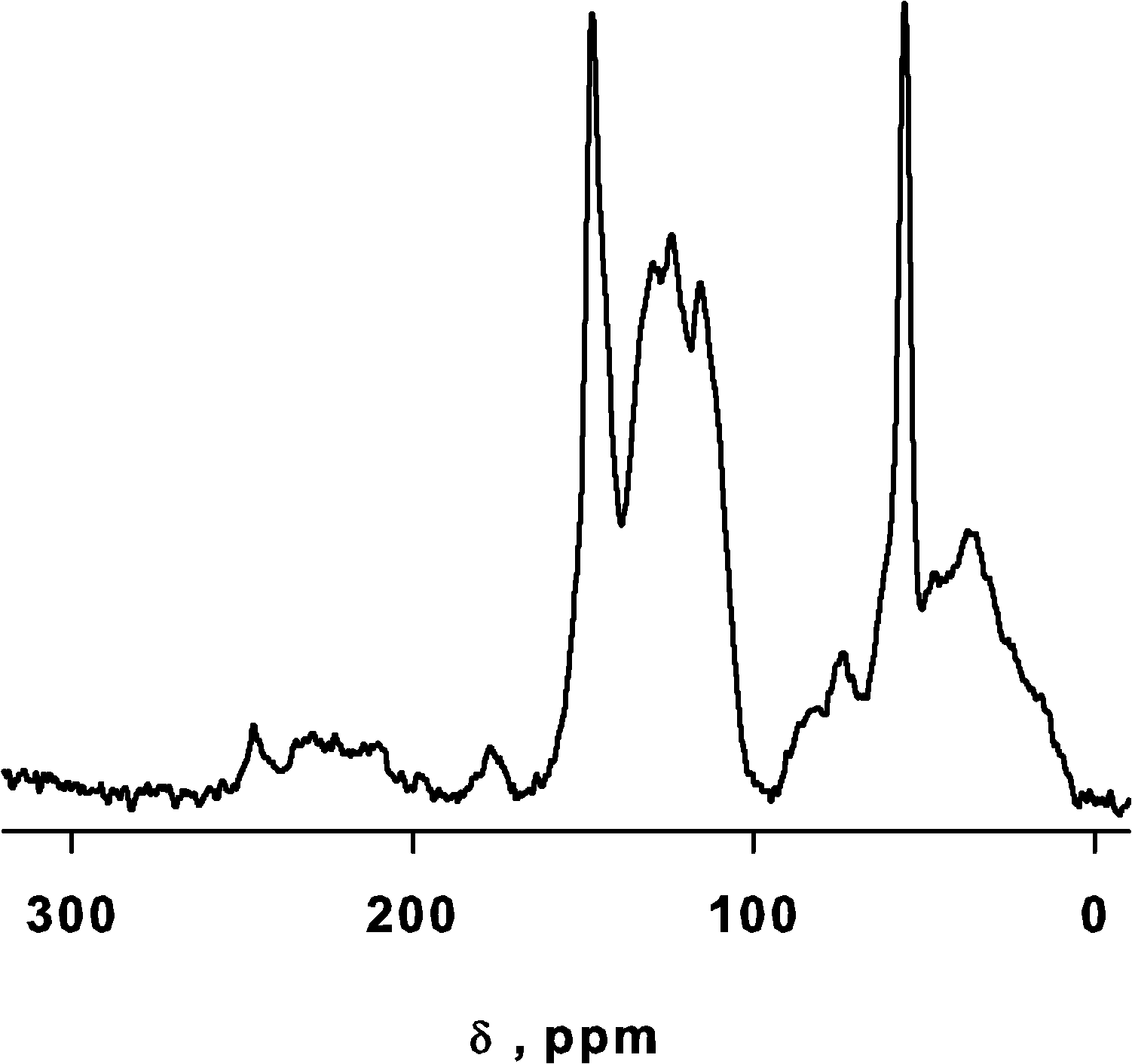 | ||
| Fig. 1 13C solid NMR spectrometer of Lignin. | ||
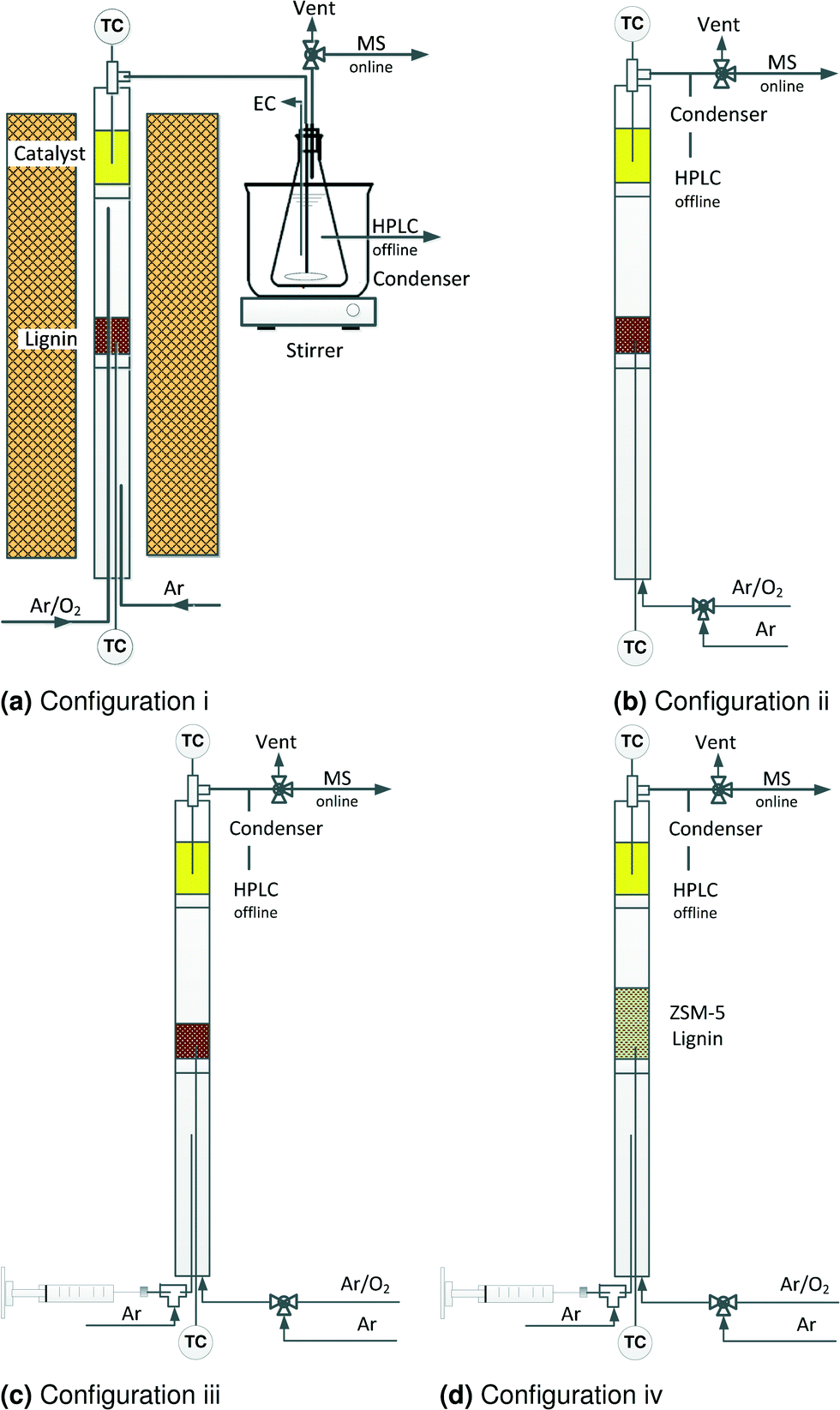 | ||
| Fig. 2 Schematic of experimental setup. | ||
• C1: V–Mo/Al2O3 (ref. 21)
• C2: V–Mo/TiO2
• C3: V2O5/MnO2
• C4: MgO (Sigma-Aldrich—we did not purify it further)
• C5: Mg–K (Pingxiang Xingfeng Chemical Packing Co., Ltd31)
• C6: WO3/TiO2 (ref. 32 and 33)
• C7: V–Mo/ZSM-5
• C8: V–W/ZSM-5
C1: We impregnated γ-alumina (specific surface area (SSA) = 140) with V–Mo. The details are described in ref. 21.
C2: We impregnated a commercial TiO2-IV with the metal salts and followed the same procedure as in C1 but we replaced Al2O3 with TiO2.
C3: To synthesize V2O5/MnO2 we added 1 g of MnO2-IV powder to the aqueous ammonium metavanadate (1.3 g) and ammonium hydroxide (0.07 g) solution at 80 °C.34 After impregnation, a rotary evaporator removed most of the water at 500 mmHg and 85 °C. The powder dried at 100 °C and atmospheric pressure for 12 h. A static air muffle furnace calcined the V2O5/MnO2 sample at 500 °C for 6 h.
C7: V and Mo salts impregnated HZSM-5 according to the procedure C1.
C8: For V–W/ZSM-5 we impregnated HZSM-5 with salts of V and W: 10% W and 30% V.
The catalyst reagents included: HCl (Fischer Scientific, 37%), ammonium molybdate(VI) tetrahydrate (Acros, 99%), sodium tetraborate decahydrate (Fisher Scientific, 99.5%), sodium phosphate (Sigma-Aldrich,96%), ammonium metavanadate (ACS reagent grade, ≥99%), V2O5 (99.6%). The supports were HZSM-5 (Alfa Aesar, SiO2/Al2O3 molar ratio 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), titanium(IV) oxide (Alfa Aesar, 99.5%-metals basis), activated aluminum oxide (ACROS), manganese(IV) oxide and magnesium oxide (Sigma-Aldrich). We did not further purify any reagents.
:1), titanium(IV) oxide (Alfa Aesar, 99.5%-metals basis), activated aluminum oxide (ACROS), manganese(IV) oxide and magnesium oxide (Sigma-Aldrich). We did not further purify any reagents.
2.2 Catalyst characterization
A Quantachram Autosorb 1 MP porosimeter measured the surface area and porosity with 21 points, following the standard Brunauer–Emmet–Teller (BET) method. Prior to the test, a heated bag degassed the samples at 200 °C for 3 h under vacuum.A Philipps X'pert diffractometer with a Cu-K radiation source (1.5406 Å) at 50 kV and 40 mA generated the diffraction patterns (XRD). The instrument operated at room temperature with an angle of incidence of 0.5°. It scanned diffraction angles between 20° and 90° with a divergence slit of 1.
A scanning electron microscope (SEM) generated images of the catalysts. An SEM-JSM-840A (JOEL Company) energy-dispersive X-ray spectroscope (SEM-EDX) coupled with an electronic X-ray diffraction (EDX) detector identified the principal elements in the catalytic samples.
A Perkin Elmer infrared spectrometer (FTIR) with a spectral resolution of 4 cm−1 scanned the lignin and reactor's residue at 16 kHz. The data were collected in the range from 4000 cm−1 to 600 cm−1. The surface composition of the samples was investigated using attenuated total reflectance (ATR) mode.
2.3 Reactor configurations
In a first set of experiments, vanadium pyrophosphate (VPP) catalyst converted lignin to carboxylic acids in four reactor configurations in two steps. During the pyrolysis and thermo-oxidation of lignin, monomers are among the first products but they react further to form carbon-oxides or re-polymerize during thermo-oxidation and pyrolysis consequently. Selective oxidation of these monomers to chemicals instead of gas or solid (coke) could improve product selectivity. Accordingly, we devised four two-step reactor configurations. In the first step, lignin decomposed to volatile compounds. These compounds reacted with the heterogeneous catalysts in the second step.• i) Lignin pyrolysis + oxidation over VPP (Fig. 2a)
• ii) Lignin thermo-oxidation + oxidation over VPP (Fig. 2b)
• iii) Lignin thermo-oxidative steamcracking + oxidation over VPP (Fig. 2c)
• iv) Lignin thermo-oxidative steamcracking over V–Mo/HZSM-5 + oxidation over VPP (Fig. 2d)
Reactor configuration iii maximized the yield of carboxylic acids and therefore we selected it to assess catalyst performance—yield, conversion, selectivity, product profile – in the second set of experiments (Fig. 2c). The reactor was an 8 mm ID quartz tube 600 mm long with two glass wool distributors positioned 350 mm and 500 mm from the bottom. Placing the catalyst bed near the top of the reactor minimizes product degradation.
In all the experiments we loaded 250 mg of lignin above the lower distributor and 500 mg catalyst above the top distributor. The gas contact time in the catalyst bed was 0.2 s.
Two thermocouples monitored the temperature in each zone. A syringe pump injected water into the reactor from the bottom through a 0.15 mm nozzle. Argon entered the nozzle at 22 mL min−1 to help atomize it into small droplets.
The mole fraction of oxygen in the feed bottle was 33% (balance Ar) at a flowrate of 38 mL min−1. The temperature ramp for all experiments was:
• 15 °C min−1 up to 150 °C
• 5 °C min−1 up to 350 °C
• 5 °C min−1 up to 450 °C, (75 min from the beginning of the reaction)
• 5 °C min−1 up to 550 °C (105 min from the beginning of the reaction)
The choice of the heating rate was dictated by TGA experiments in air, which showed that lignin lost ∼40% of its weight above ∼500 °C. We repeated selected tests two or three times.
A Varian HPLC (Metacarb 87H column) analyzed the liquid sample offline. We calibrated the HPLC with standard solutions for each product we detected, building a calibration curve with six concentrations. We repeated each analysis three times. A Pfeiffer mass spectrometer (MS) monitored the permanent gases online.
At the end of the last temperature ramp and hold at 550 °C no solids remained above the first distributor. We calibrated the MS with a mixture of hydrocarbons and oxygen at four concentrations. The carbon balance was the sum of the carbon in the gas phase (MS) + the carbon in the liquid phase + the carbon remained on the catalyst surface. The mass of carbon remaining on the catalyst we measured by TGA. The MS detected CO, CO2 and CH4. We calculated the carbon trapped in the water-ice quench as the difference between the carbon loaded to the reactor and that detected on the catalyst and in the gas phase and by HPLC. We assumed that the response factors for the unknown aromatics (detected by HPLC) were the same as other known aromatics that we had previously evaluated by GC-MS.21 These two methods differed by less than 10%.
3 Results and discussions
3.1 Two-step lignin degradation
We proposed a reaction pathway to from maleic acid from lignin: the V5+ ion activates lignin to attract p electrons of the double bonds and generate a positive charge displaced on the ring. The ring opens by successive attack of 2O2.21
Here, 19% of the carbon in the lignin converted to liquid. The selectivity to maleic acid was 84%. The production of light monomers during pyrolysis could account for the high maleic acid selectivity. Other products were aromatics (6%) acrylic acid (2.6%), malonic acid (2.6%), followed by formic, lactic, acetic acid and vanillin. The solid residue remaining above the first distributor was the highest among the four configurations (46%) due to char (Table 1).
| Name | Config. i | Config. ii | Config. iii | Config. iv |
|---|---|---|---|---|
| Carbon balance (%) | ||||
| Liquid | 19 | 23 | 24 | 13 |
| Solid | 46 | 4 | 5 | 6 |
| Gas | 35 | 73 | 71 | 81 |
| Liquid selectivity, (%) | ||||
| Aromatic | 6 | 36 | 31 | 16 |
| Maleic/fumaric acid | 84 | 36.5 | 48 | 48 |
| Butyric acid | — | — | — | 10 |
| Malonic acid | 3.5 | 6 | 2.7 | 12 |
| Formic acid | 1 | 0.5 | 0.65 | 0.5 |
| Acetic acid | 0.1 | 0.3 | — | 1 |
| Lactic acid | 0.2 | 0.8 | 0.8 | — |
| Phthalic acid | 0.01 | 0.3 | 0.1 | 7 |
| Vanillin | ✓ | 0.3 | — | 2.6 |
| Gallic acid | 0.7 | 1.5 | 1.2 | — |
| Acrylic acid | 2.6 | 3 | 7 | — |
| Syringic acid | 0.06 | — | — | — |
| Unknown | 0.8 | 13.5 | 7 | 1.5 |
| Benzoquinone | 0.2 | 1 | 1.2 | 0.6 |
| H2 (%) | 0.6 | 0.2 | 1.3 | 0.7 |
We attribute the large gas fraction produced in this configuration (73%) to either the decarbonylation of lignin monomeric units, lignin gasification, or to combustion of coke on the catalyst to CO and CO2 (Table 1).
The high cracking activity of V–Mo/HZSM-5 is responsible for the high yield of gases, as well as the combustion of the coke on the catalyst during the reaction.
Beside lignin, we tested unwashed lignin in the same condition as conf. iv. 19%, 76%, 5% of carbon converted to liquid, gas and solid, respectively. MA selectivity reached up to 68% and we detected 9% aromatic, 5% malonic, 1.5% phthalic and formic acid.
Fig. 3a FTIR spectra shows the typical lignin derivatives. Adsorption at 3420 cm−1, 2944 cm−1 and 2833 cm−1 corresponds to the stretching of O–H, aromatic C–H and methoxyl groups, respectively. The bands in the range 3000 cm−1 to 2860 cm−1 correspond to the aliphatic C–H stretching, whereas the ones above 3000 cm−1 to the C–H aromatic stretching, which overlaps the band of O–H. We attribute the band at 1711 cm−1 to the carbonyl group of aldheydes. The bands at 1520 cm−1 and 1594 cm−1 are typical of skeletal vibrations of the aromatic ring. The higher intensity of the band at 1520 cm−1 compared to the one at 1594 cm−1 is typical of softwood lignin.38 C–H deformation and aromatic ring vibrations also adsorb at 1464 cm−1 and 1408 cm−1. We attribute the bands at 1268 cm−1 and 1222 cm−1 to the C–O bonds in syringyl and guaiacyl units and the bands at 1138 cm−1 and 1082 cm−1 to the in-plane aromatic C–H deformation typical of the same compounds.39 The band at 1036 cm−1 refers to the C–O stretch of the O–CH3 and C–OH. The bands at 868 cm−1 and 822 cm−1, respectively, may correspond to lone aryl CH wag and two-adjacent aryl CH wag, respectively. The band at 626 cm−1 may represent the out of plane O–H band.
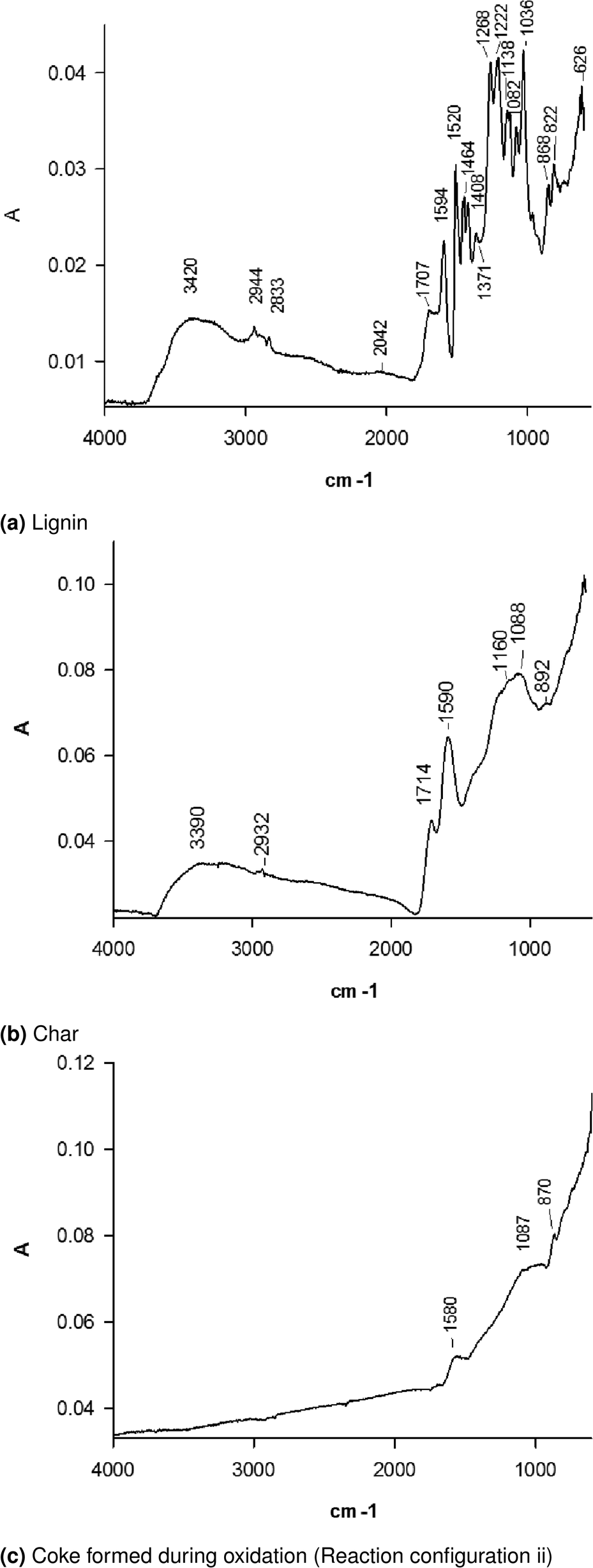 | ||
| Fig. 3 FTIR spectrum. | ||
In upward shift of the FTIR spectrum baseline for most of the peaks of char remaining on the lower distributor may relate to the large carbonized compared to lignin (Fig. 3b). Many typical absorption peaks of lignin disappear, in particular, the signals of the aliphatic groups. The OH-stretching band at 3390 cm−1 represent phenolic hydroxyl group.38 C–O in carbonyl group or in carbonyl compounds conjugated with aromatic ring (1714 cm−1) and aromatic ring (1590 cm−1) are still present. The bands at 1160 cm−1 and 1088 cm−1 are typical of the in-plane aromatic C–H deformation of aromatic syringyl and guaiacyl units.39
The FTIR spectrum of coke on V–Mo/HZSM-5 after the lignin degradation in reactor configuration iv has a typical band at ∼1580 cm−1 (Fig. 3c). This band belongs to the highly unsaturated carbonaceous deposit called “hard coke”. The bands at 1087 cm−1 and 870 cm−1 that are typical of the in-plane aromatic C–H deformation of aromatic syringyl and guaiacyl units persist.39
3.2 Thermo-oxidative steam cracking: catalytic activity
Besides VPP, we tested 8 other catalysts to identify the most selective towards carboxylic acids (Table 2). Metal oxides of group V and VI, like V, Mo, and W are common catalysts to partially oxidize hydrocarbons, particularly vanadium.40,41 Phosphoric, arsenic and boric acids control the activity of V, Mo and W in the gas phase oxidation of hydrocarbons.42| Name | C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 |
|---|---|---|---|---|---|---|---|---|
| Catalyst | V–Mo/Al2O3 | V–Mo/TiO2 | V2O5/MnO2 | MgO | Mg–Si–Al–K cat. | WO3/TiO2 | V–Mo/HZSM-5 | V–W/HZSM-5 |
| EC (μS cm−1) | — | 103 | 107 | 12 | 118 | 174 | 155 | 130 |
| Carbon balance (%) | ||||||||
| Liquid | 23 | 15.5 | 16.5 | 8.5 | 19.5 | 20 | 25 | 21 |
| Solid | 6.5 | 4.5 | 3.5 | 11 | 6 | 4 | 4 | 7.5 |
| Gas | 70.5 | 80 | 80 | 80.5 | 74.5 | 76 | 71 | 71.5 |
| Liquid selectivity (%) | ||||||||
| Aromatic | 64 | 42 | 36 | 7 | 33.2 | 11 | 19 | 9.5 |
| Maleic/fumaric acid | 20.5 | 45 | 18.5 | 1 | 1.5 | 9 | 26 | 56 |
| Butyric acid | 0.6 | 0.5 | 16 | 11 | 32.5 | 64 | 17 | 14 |
| Malonic acid | — | — | — | — | ✓ | 0.4 | — | — |
| Formic acid | 1 | 0.7 | — | — | 0.06 | ✓ | — | 0.1 |
| Acetic acid | 0.2 | 0.6 | — | — | 0.2 | 0.1 | — | 0.1 |
| Lactic acid | 5 | — | — | — | — | — | — | |
| Muconic acid | — | — | 1.5 | — | — | — | ||
| Succinic acid | 0.8 | 7.5 | 3 | — | — | — | 1 | 8.5 |
| Phthalic acid | 0.3 | 2 | 2.5 | 7 | 7.5 | 5 | 0.5 | 1 |
| Vanillin | ✓ | 0.5 | 0.3 | 11 | 1.5 | 1.5 | 5 | — |
| Benzoic acid | — | — | 0.1 | 2.5 | 15 | 1.5 | 8 | — |
| Gallic acid | 0.3 | — | 0.15 | — | ✓ | ✓ | — | 0.3 |
| Acrylic acid | 5.5 | — | 1 | — | — | — | — | — |
| Syringic acid | 0.6 | — | — | — | — | — | — | — |
| Unknown | 0.7 | 0.7 | 20 | 59.5 | 8 | 7 | 23.1 | 10.2 |
| Benzoquinone | 0.6 | — | 0.2 | 1 | — | — | 0.3 | 0.2 |
| H2 (%) | 2 | — | 7 | — | 3.3 | 4 | 5.5 | 6 |
Neto et al.40 designed a multi-metal oxide catalyst containing (Aga−cQbMcV2Od·eH2O) to partially oxidize aromatic hydrocarbons in the gas phase to produce aldehydes, and carboxylic acids or anhydrides in fixed bed reactors.
V included in polyoxometalates catalysts degraded lignin to monomeric compounds. The catalysts comprised anionic clusters of d0 metal cations, mostly, WVI, WMoVI, VV, NbV and oxygen anions arranged in Mo6 octahedral units. Polymetalates cleave β-O-4 and C–C linkages of lignin and directly produce low molecular weight phenolic compounds in different oxidative environments and liquid solutions. As much as 90% of the lignin model compounds reacted.5 The reduction of vanadium changes with the supports and decreases as follows: γ-Al2O3 > SiO2–Al2O3 > SiO2 > α-Al2O3.43
The properties of the support oxide affect the activity and the selectivity of the supported metal significantly.44 Choosing a support is as important as the choosing the oxidation catalyst.
A base such as NaOH with an extra nucleophile like NaHS, anthraquinone, acid-catalyzed hydrolysis and hydrochloric acid or AlCl3 in dioxane–water or ethanol–water will all β-O-4 ether linkages. Lignin C–C bonds are among the most difficult bonds to break and they can also form during lignin treatment processes. The bond is broken by fluid catalytic cracking with optimized zeolites in acid-catalyzed reactions.45 The presence of sulfur in kraft lignin is a barrier in downstream catalytic processes.12
Though VO2P2O7 is unequivocally the bulk compound for what we call VPP, 15 different crystalline phases may form before or during the reaction.46 The acid sites cleave β-O-4 and α ether linkages47 and V complexes selectively cleave C–O and C–C bonds and oxidize lignin to monomeric phenolic compounds including alcohols, aldehydes, ketones, and carboxylic acids.5 In addition, their redox properties make them excellent catalysts for gas phase partial oxidations. Chieregato reports that in a O2-rich environment, the O2 atoms re-arrange on the VPP surface into finite δ-VOPO4 domains that are the selective oxidation phase.5 We chose vanadium oxides because they carry both strong Lewis acid and redox functionalities to cleave lignin bonds and partially oxidize it to carboxylic acids.
Ni, Co, Mo on Al2O3 and TiO2 degrade lignin by oxidation, hydrogenation and hydrodeoxygenation. Commercially, catalysts combining V2O5 and MoO2 partially oxidize benzene and n-butane to MA.48 The unique catalytic properties of V and Mo is the reason why they are incorporated together in catalysts for commercial processes to oxidise and partially oxidise a wide range of compounds, including acrolein, alkanes and aromatics. We selected Mo as an active species in conjunction with V because V cycles between V4+ and V5+, Mo cycles between Mo5+ and Mo6+ oxidation states. Mo5+ and Mo6+ are strong Lewis acids, as well as V4+ and V5+ that can therefore break lignin bonds and partially oxidize the intermediates. The V and Mo operate synergistically.46 Differently from V and Mo, W isn't a typical oxidation catalyst, but rather promotes hydration and dehydration reactions.33 The mechanism of lignin degration to acrylic acid and lactic acid involves hydration and de-hydration. For this reason, in C8, we supported W and V on HZSM-5.
Here, V–Mo/Al2O3 converted 23% of the lignin carbon to liquids (2). The main products in the liquid were C5–C8 aromatic compounds (S = 64%).
The second most abundant product in the quench was maleic acid (S = 20%), followed by acrylic acid (S = 5.5%), and lactic acid (S = 5%). Vanadium and VPP in particular has a unique capacity to oxidize aromatics to maleic anhydride/acid from aromatics.21,49 Jongerius report that Mo reduces catalyst deactivation and improves the cleavage of the diphenylmethane bonds.50 While the bonds involving monophenolic units split at temperatures up to 450 °C, both with and without oxygen, Mo has a role in cracking biphenyl bonds and yield phenol and benzene.51 V–Mo based catalysts oxidize o-xylene and benzene and olefins to anhydrides.52 Mo reduces the catalyst deactivation rate and increases cleavage of 4-hydroxydiphenyl ether, diphenyl ether, and diphenylmethane bonds.50 Fumagalli reports that V–Mo catalysts have a high oxygen insertion capacity that oxidize the intermediates involved in the process o-xylene to phtalic anhydride.53
We attribute the higher selectivity to maleic acid compared to lactic acid to the higher oxygen concentration with respect to our previous data 11% vs. 4% in ref. 21. Products such as formic, acetic and lactic acid form from the β-O-4 bonds cleavage at temperatures higher than 200 °C, depending on the reactivity of the substituents of the aromatic ring. The catalyst in the second stage may completely oxidize some of these acids to CO2 and H2O. We were unable to quantify how much H2O the reaction produced because of the large excess of steam we co-fed with the O2.
In our case, replacing γ-Al2O3 with TiO2 as a support decreased the yield of liquid to 16%, but increased the selectivity to maleic acid (45%). Other products were: aromatics (42%), succinic acid (7.5%), phthalic acid (7.5%), formic and acetic acid and vanillin.
Surprisingly, HZSM-5 as a support for V and Mo gave the highest selectivity to liquid products not only among the V–Mo based samples, but among all the catalysts tested. HZSM-5 zeolite is a commercial catalyst for the FCC of lineal olefins and alkanes hydrocracking.57 HZSM-5 is a strong Brønsted acid, which is desirable for the conversion of benzene to ethylbenzene and pyrolysis of biomass to liquid.2,58
A hypothesis that explains the high yield of liquids relates to the very narrow pores of HZSM-5, which might be too small to host the sterically-hindered intermediates of the lignin steam cracking derived from the first stage. The narrow pores prevent the production of those reaction intermediates that are the precursors of coke.58 Nevertheless, the selectivity to aromatics is lower (19%) compared to C1 and C2 and maleic acid (+fumaric acid) and butyric acid accounts for almost 40% of the selectivity to liquids. Other products are benzoic acid (8%), vanillin and phthalic acid.
WO3 has strong Lewis acid sites (W6+)54,67 that can activate the intermediates deriving from the lignin degradation of the first reaction stage (see the next section of reaction pathways).
| Catalyst | A m2 g−1 | Pore volume cm3 g−1 |
|---|---|---|
| V–Mo/Al2O3 | 115 | 0.25 |
| V–Mo/TiO2 | 53 | — |
| VPP | 41 | 0.074 |
| V–Mo/ZSM–5 | 46 | 0.095 |
| V–W/ZSM–5 | 50.7 | 0.124 |
The diffractogram of V–Mo/Al2O3 was typical of an amorphous material. The XRD pattern of V–Mo/Al2O3 and V–Mo/TiO2 (Fig. 4) resemble the data of Shishido et al.68 Orthorhombic Mo4O11 crystallites and V2O3 form on TiO2.68 Peaks at 2θ = 24, 27, 33, 36, 37, 55 and 57 relates to V2O3 phase and peaks at 2θ = 21.8, 26, and 33.4 belongs to Mo4O11 (JCPDS 13-042).
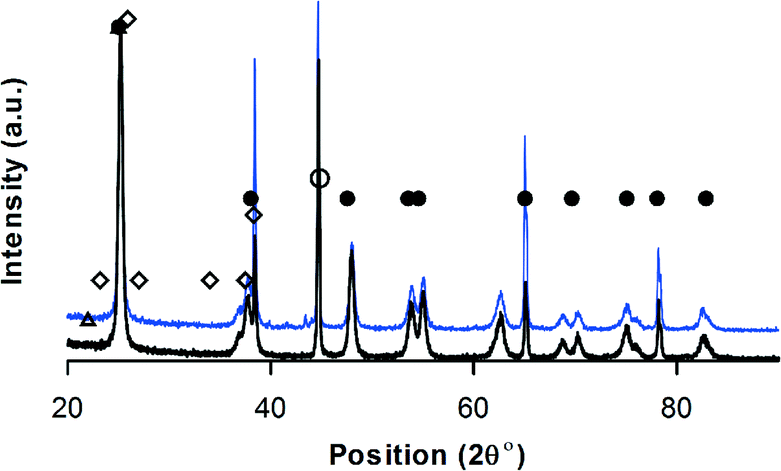 | ||
| Fig. 4 XRD-V–Mo/TiO2, • anatase TiO2, ⋄ MoO3, △ Mo4V6O25, ∘ VO2,69 black = before reaction – blue = after reaction. | ||
Triclinic aluminum vanadium oxide (AlVO4), tetragonal vanadyl molybdenum oxide (VOMoO4), and monoclinic molybdenum vanadium oxide (Mo0.67V0.33O2) forms on Al2O3 (Fig. 4).68
(VO)2P2O12, (VO)2P2O7, VO(PO3)2 and VOHPO4·0.5H2O phases constitute VPP (Fig. 5). As expected, (VO)2P2O7 is the predominant phase, which is responsible for the catalytic activity in oxidation. VOHPO4·0.5H2O, which is the surface product of O2 rearrangement in the lattice is also present.21
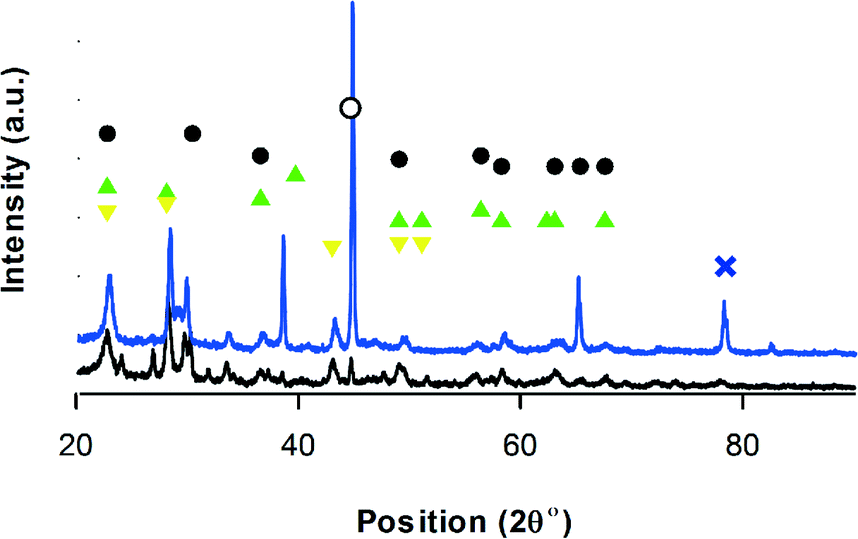 | ||
Fig. 5 XRD-VPP, • (VO)2P2O7,70 VO(PO3)2, VO(PO3)2,  VOHPO4·0.5H2O, ∘ VO2,69 VOHPO4·0.5H2O, ∘ VO2,69 V6O13,71 black = before reaction – blue = after reaction. V6O13,71 black = before reaction – blue = after reaction. | ||
V2O5 (2θ = 20, 22, 26, 31, 32, 33, 34, 39.5, 48, 52, 55, 58) and MoO3 (2θ = 24, 26, 27, 34, 38 and 39) phases forms on HZSM-5.72,73 XRD detected VMoO8 and V0.07Mo0.93 on HZSM-5 (Fig. 6). These are the active V–Mo mixed oxides for oxidation reactions.46
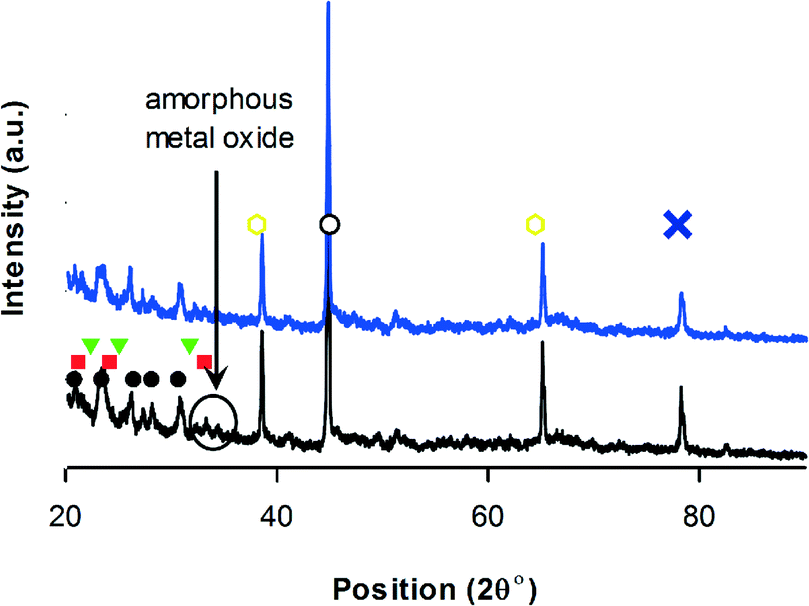 | ||
Fig. 6 XRD-V–Mo/ZSM-5, • HZSM-5,74 VMoO8, VMoO8,  (V0.07Mo0.93)5O14, ∘ VO2,69 (V0.07Mo0.93)5O14, ∘ VO2,69 V6O13,71 MoO3,75 black = before reaction – blue = after reaction.76 V6O13,71 MoO3,75 black = before reaction – blue = after reaction.76 | ||
After the reaction, the phases detected in V–Mo/TiO2 and V–Mo/ZSM-5 were the same as for the fresh catalyst.77 In VPP, the intensity of the peaks at ∼30 and 42 reduced. This peak belongs to monoclinic VO2 and is observed in all XRD diffractogram of all the catalysts (Fig. 4–6).69
3.3 Discussion
 | ||
| Fig. 7 WO3/TiO2. | ||
Aromatic compounds. Steam and oxygen crack the lignin in the void space below the upper distributor. We hypothesize that in this first step the β-O-4 bonds cleave and release the aromatic phenolic units that constitute the monomers of lignin. These units pass through the bed above the lignin or react with the catalyst to form carboxylic acids (2). The reactivity of the monomeric aromatic units depends on the type of substituents carrying oxygen functionalities.19 A GC-MS detected aromatic compounds for all the catalysts tested, in particular for the samples C1, C2, C3, C5. 1-Ethyl-3-methyl cyclopentane, cyclopentane 1-ethyl-1-methyl, cyclohexane-1,4 dimethyl, ethyl benzene, benzene, 1,3-dimethyl, cyclohexane methyl, and others formed the aromatics.
Maleic and fumaric acid. Recently, we derived a mechanism to describe how maleic anhydride, lactic acid, acetic acid and formic acid (and phthalic anhydride) form from pure lignin.21 Vanadium has a unique capacity to produce maleic anhydride/acid. V5+ activates the aromatic rings of the monomeric units increasing their electrophylicity, thus exposing them to the attack of 2 molecules of O2. After the attack of 2 O2 the ring opens and re-arranges to maleic anhydride. Maleic anhydride hydrates to maleic acid and its isomer fumaric acid.
Butyric acid. Here for the first time we report a mechanism to account for the high concentration of butyric acid (Fig. 8). We took as a representative catalyst WO3/TiO2. We hypothesize that butyric acid (and crotonic acid) form from lignin in 3 steps. Step 1 includes the oxidative steam cracking of the lignin. At temperatures above 200 °C the β-O-4 bonds cleave and release the aromatic phenolic units in the reactor space above the lignin bed below the upper distributor. This rupture generates either an anion (phenolate) or a phenol radical. The negative charge or the radical will most likely displace to carbon atoms of the ring that carry an oxygen functionality to give the chetonic form.78 In this form the carbon of the carbonyl group is strongly electrophilic and will undergo nucleophilic attack by water (steam cracking). In the attempt to recover its aromaticity, the rings open. Gierer et al. observed the same type of aromatic ring cleavage in liquid phase and presence of (H2O2) under both alkaline and acid conditions.78 During step 1, O2 might oxidize the constituents of the aromatic rings, producing carboxylic acids.21
 | ||
| Fig. 8 Proposed Mechanism of formation of butyric acid in 3 steps. | ||
Step 2 includes the activation of the intermediates formed by ring cleavage onto the catalyst. We hypothesize that the acid sites on the catalyst, either of Brønsted or Lewis type, are the active species involved in this step. WO3 carries both strong Brønsted and Lewis acid sites. Lewis acid sites are W6+.54,67 V5+ is a Lewis acid strong enough to activate the reaction intermediate formed during step 1. In the activation mechanism the positive charge of tungsten displaces the p electrons of the open aromatic ring, similarly as V5+ activates the aromatic ring after β-O-4 bonds cleavage.21 O2 adds to the intermediates giving hydroxyls. The intermediate cleaves when an excess of electrons accumulate around an electrophilic carbon.
Step 3 involves hydrogen. We hypothesize that the intermediate formed during step 2 remains anchored to tungsten until hydrogen adds to it. Until the intermediate is absorbed on W it can be oxidized up to carboxylic acid, but only H2 can displace it from the metal. Butyric acid forms after dehydration followed by hydrogenation of the crotonic acid.
Karlsson et al.79,80 report butyric acid derives from the ozonolysis of lignin-carbohydrate complex model compound, i.e. in strong oxidative conditions. Ozonolysis of β-O-4 structures produces erythronic and threonic acids,81 which are precursors of the butanoic acid. Shao et al. report that GC-MS analyses detected butanoic acid both in a untreated lignosulfonate sample and after oxidation above Ti/SbASnO2 and Ti/PbO2 electrodes.82
We hypothesize that butyric acid could form from the condensation of two molecules of acetaldehyde followed the dehydration over WO3/TiO2 and successive oxidation to the acid. We tested this hypothesis and fed acetaldehyde over the catalyst under the same conditions but only detected acetic acid in the quench.
Lactic and malonic acids. Acrylic acid hydrates to form lactic acid.21 However, another possible mechanism to account for lactic acid as well as malonic acid involves cleaving propylic chains of monomeric units such as syringyl and guaiacyl (Fig. 9).
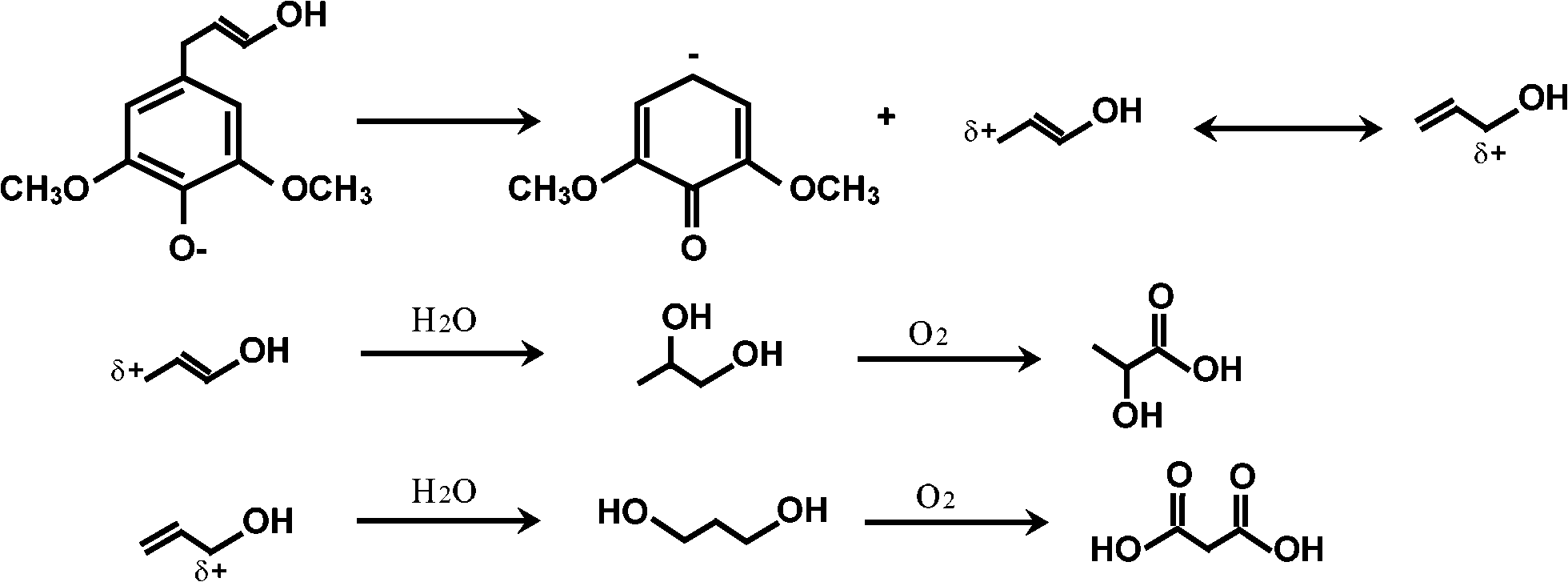 | ||
| Fig. 9 Proposed mechanism of formation of lactic and malonic acids from syringyl alcohol. | ||
Formic and acetic acid. The cleavage of methyl and ethyl constituents followed by oxidation produces formic and acetic acids.21
Acrylic acid. Acrylic acid may form from the scission of a phenolic unit and one of the terminal aldehydes as a consequence of the dehydration followed by oxidation.21 At high temperatures, lactic acid may dehydrate to acrylic acid.
Muconic acid. Muconic acid is a C6 carboxylic acid that can form from the aromatic ring opening, followed by oxidation and dehydration.83
Succinic acid. The MS detected H2 in the gas phase which could hydrogenate maleic and fumaric acids to succinic acid. The hydrogen may form either from the gasification of lignin, which decomposes into elements (H2 and C forming coke on the catalyst)21,84 or from steam reforming of the biomass into syngas (H2 + CO). The concentration of succinic acid follows the same trend as that for maleic acid for the eight catalysts (Table 2): V–Mo/TiO2 and V–W/HZSM-5 both produce the most maleic and succinic acids whereas the MgO and MgSiAlK make little maleic and no succinic acid.
4 Conclusions
Lignin is an inert macromolecule and because of its heterogeneity remains underexploited commercially. Activating it with high temperatures (as in pyrolysis), oxygen and/or water vapour produces phenolic compounds and bio-oils. Catalysts can improve the selectivity to target compounds and effectively decreases the required operating temperature. Pre-mixing the lignin with catalyst increases the cracking rate but it produces more coke, char and gas. Moreover, the catalyst deactivates more rapidly. We demonstrated a two-step process in which lignin is thermo-oxidatively steam cracked in the first step to volatile compounds which contact a catalyst bed in the second step. Little coke formed on the catalyst (less than 5% of the total C in lignin) and the catalyst did not agglomerate.V–Mo/Al2O3 and V–Mo/HZSM-5 converted 23% and 25% of the lignin into liquid products, respectively. The selectivity to maleic acid was 20% in both cases. Replacing Al2O3 and HZSM-5 with TiO2 reduced the liquid yield (15.5%) but increased maleic acid selectivity (45%). Replacing molybdenum with tungsten in V–Mo/HZSM-5 produced, butyric acid and succinic acid together with maleic acid.
Acknowledgements
The authors acknowledge NSERC for financial support and DuPont for supplying the VPP catalyst and FPinnovation for supplying lignin and analytical assistance. In addition, special thanks to NSERC Biomaterials and Chemicals Strategic Network (http://www.lignowork.ca). We acknowledge NSERC for the Banting post-doctoral fellowship granted to Dr. Boffito. We wish to thank Dr. Amir Saffar from Polytechnique de Montréal for helping with the FTIR analysis.References
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- M. Fan, P. Jiang, P. Bi, S. Deng, L. Yan, Q. Zhai, T. Wang and Q. Li, Bioresour. Technol., 2013, 143, 59–67 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- D. Lalitendu, K. Praveen and S.-S. Ratna, Biofuels, 2012, 3, 155–166 CrossRef.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmannb and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 CAS.
- E. Feghali, G. Carrot, P. Thuery, C. Genre and T. Cantat, Energy Environ. Sci., 2015, 8, 2734–2743 CAS.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- A. Witsuthammakul and T. Sooknoi, Appl. Catal., A, 2012, 413–414, 109–116 CrossRef CAS.
- X. Xu, J. Lin and P. Cen, Chin. J. Chem. Eng., 2006, 14, 419–427 CrossRef CAS.
- K. Kadowaki, K. Sarumaru and T. Shibano, Production of acrylic acid US3773828, 1980 Search PubMed.
- M. Kwan, N. Cant, D. Trimm and M. Wainwright, Appl. Catal., 1987, 31, 25–34 CrossRef CAS.
- Y. Ma, Z. Du, J. Liu, F. Xiaab and J. Xu, Green Chem., 2015, 17, 4968–4973 RSC.
- C. S. Lancefield and N. J. Westwood, Green Chem., 2015, 17, 4980–4990 RSC.
- M. Brebu and C. Vasile, Cellul. Chem. Technol., 2010, 44, 353–363 CAS.
- R. N. Olcese, G. Lardier, M. Bettahar, J. Ghanbaja, S. Fontana, V. Carré, F. Aubriet, D. Petitjean and A. Dufour, ChemSusChem, 2013, 6, 1490–1499 CrossRef CAS PubMed.
- S. Lotfi, D. C. Boffito and G. S. Patience, ChemSusChem, 2015, 8, 3424–3432 CrossRef CAS PubMed.
- W. Mu, H. Ben, A. Ragauskas and Y. Deng, BioEnergy Res., 2013, 6, 1183–1204 CrossRef CAS.
- L. Kouisni, P. Holt-Hindle, K. Maki and M. Paleologou, J. Sci. Technol. For. Prod. Processes, 2012, 2, 6–10 Search PubMed.
- L. Kouisni and M. Paleologou, Method for separating lignin from black liquor, US Pat., 8771464, 2014 Search PubMed.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5, 87187–87199 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- L. V. Kanitskaya, A. F. Gogotov, D. T. T. Khai and A. V. Rokhin, Russ. J. Bioorg. Chem., 2012, 38, 720–725 CrossRef CAS.
- L. B. Davin, M. Jourdes, A. M. Patten, K.-W. Kim, D. G. Vassao and N. G. Lewis, Nat. Prod. Rep., 2008, 25, 1015–1090 RSC.
- Y. Pu, B. Hallac and A. J. Ragauskas, Plant Biomass Characterization: Application of Solution- and Solid-State NMR Spectroscopy, John Wiley Sons, Ltd, 2013, pp. 369–390 Search PubMed.
- G. S. Patience and R. E. Bockrath, Appl. Catal., A, 2010, 376, 4–12 CrossRef CAS.
- Pingxiang XingFeng Chemical Packing Co., Ltd.
- J. Dubois, C. Duquenne and W. Hoelderlich, Process for dehydrating glycerol to acrolein, WO Pat. App. PCT/EP2006/000,735, 2006 Search PubMed.
- M. Dalil, D. Carnevali, J.-L. Dubois and G. S. Patience, Chem. Eng. J., 2015, 270, 557–563 CrossRef CAS.
- V. Lochar and L. Smoláková, React. Kinet. Catal. Lett., 2009, 96, 117–123 CrossRef CAS.
- M. J. Lorences, G. S. Patience, F. V. Díez and J. Coca, Ind. Eng. Chem. Res., 2003, 42, 6730–6742 CrossRef CAS.
- S. Farag, L. Kouisni and J. Chaouki, Energy Fuels, 2014, 28, 1406–1417 CrossRef CAS.
- M. T. Klein and P. S. Virk, Energy Fuels, 2008, 22, 2175–2182 CrossRef CAS.
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. Lin, W. G. Chan and M. R. Hajaligol, Fuel, 2004, 83, 1469–1482 CrossRef CAS.
- M. M. Ibrahim, M. N. Nadiah and H. Azian, J. Appl. Sci., 2006, 6, 292–296 CrossRef CAS.
- S. Neto, H. Hibst, F. Rosowski and S. Storck, Multimetal oxide containing silver, vanadium and a promoter metal and use thereof, US Pat., 7462727, 2008 Search PubMed.
- T. Heidemann, F. Rosowski, G. Linden, M. Seufert, G. Hefele and P. M. Lorz, Method for producing shell catalysts for the catalytic vapor-phase oxidation of aromatic hydrocarbons and catalysts obtained in such a manner, 2003 Search PubMed.
- L. Lioyd, Handbook of Industrial Catalysts, CSpringer, 2011 Search PubMed.
- M. Y. H. Kwan, Ph.D. thesis, University of New South Wales, 1985 Search PubMed.
- B. M. Weckhuysen and D. E. Keller, Catal. Today, 2003, 78, 25–46 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- A. Chieregato, J. M. L. Nieto and F. Cavani, Coord. Chem. Rev., 2015, 3–23 CrossRef CAS.
- S. Jia, B. J. Cox, X. Guo, Z. C. Zhang and J. G. Ekerdt, Hydrolytic cleavage of β-O-4 ether bonds of lignin model compounds in an ionic liquid with metal chlorides, Ind. Eng. Chem. Res., 2011, 849–855 CrossRef CAS.
- R. S. Barker, Method of oxidizing benzene to maleic anhydride using a vanadium, molybdenum, boron containing catalyst, US Pat., 3867412, 1975 Search PubMed.
- C. Uraz and S. Atalay, Chem. Eng. Technol., 2007, 12, 1708–1715 CrossRef.
- A. L. Jongerius, R. Jastrzebski, P. C. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS.
- M. Koyama, Bioresour. Technol., 1993, 44, 209–215 CrossRef CAS.
- P. F. Lovell, AIChE J., 1981, 27, 316–316 CrossRef.
- C. Fumagalli, G. Golinelli, G. Mazzoni, M. Messori, G. Stefani and F. Trifiró, in Production of Maleic and Phtalic Anhydrides by Selective Vapor Phase Oxidation with Vanadum Oxide Based Catalyst, ed. V. C. Corberán and S. V. Bellón, 1994, vol. 82, pp. 221–231 Search PubMed.
- G. Ramis, G. Busca, C. Cristiani, L. Lietti, P. Forzatti and F. Bregani, Langmuir, 1992, 8, 1744–1749 CrossRef CAS.
- G. Centi, Appl. Catal., A, 1996, 267–298 CrossRef CAS.
- S. Bagheri, N. M. Julkapli and S. B. A. Hamid, Sci. World J., 2014, 2014, 727496 Search PubMed.
- A. Corma and L. Sauvanaud, Catal. Today, 2013, 218-219, 107–114 CrossRef CAS.
- S. Kaliaguine and A. Mahay, Catalysis on the Energy Scene, Elsevier, 1984, vol. 19, pp. 93–100 Search PubMed.
- A. Akbari and S. M. Alavi, Chem. Eng. Res. Des., 2015, 286–296 CrossRef CAS.
- H. Chen, M. Tang, Z. Rui and H. Ji, Ind. Eng. Chem. Res., 1996, 267–298 Search PubMed.
- Y. Zhu, M. SHen, Y. Xia and M. Lu, Catal. Commun., 2015, 37–43 CrossRef CAS.
- H. P. Dias, G. R. Goncalves, J. C. Freitas, A. O. Gomes, E. V. R. D. Castro, B. G. Vaz, G. M. F. V. Aquije and W. Romao, Energy Convers. Manage., 2014, 326–333 Search PubMed.
- Z. D. Yigezu and K. Muthukumar, Fuel, 2015, 113–121 Search PubMed.
- D. C. Boffito, C. Neagoe, M. Edake, B. Pastor-Ramirez and G. Patience, Catal. Today, 2014, 13–17 CrossRef CAS.
- D. C. Kannan, Ph.D. thesis, UMI, 2009 Search PubMed.
- C. Bjórgen, One-pot Conversion of Biomass to Ethylene Glycol, Propylene Glycol and other Polyols over Carbon Supported Tungsten Catalysts, 2013 Search PubMed.
- A. Gutiérrez-Alejandre, J. Ramírez and G. Busca, Langmuir, 1998, 14, 630–639 CrossRef.
- T. Shishido, A. Inoue, T. Konishi, I. Matsuura and K. Takehira, Catal. Lett., 2000, 68, 215–221 CrossRef CAS.
- L. Liu, F. Cao, T. Yao, Y. Xu, M. Zhou, B. Qu, B. Pan, C. Wu, S. Wei and Y. Xie, New J. Chem., 2012, 36, 619–625 RSC.
- E. Bordes, Catal. Today, 1987, 1, 499–526 CrossRef CAS.
- X. Wang, H. Li, Y. Fei, X. Wang, Y. Xiong, Y. Nie and K. Feng, Appl. Surf. Sci., 2001, 177, 8–14 CrossRef CAS.
- A. Teimouri, B. Najari, A. Najafi Chermahini, H. Salavati and M. Fazel-Najafabadi, RSC Adv., 2014, 4, 37679–37686 RSC.
- C. A. Chagas, L. C. Dieguez and M. Schmal, Catal. Lett., 2012, 142, 753–762 CrossRef CAS.
- B. Liu, L. France, C. Wu, Z. Jiang, V. L. Kuznetsov, H. A. Al-Megren, M. Al-Kinany, S. A. Aldrees, T. Xiao and P. P. Edwards, Chem. Sci., 2015, 6, 5152–5163 RSC.
- R. Naouel, H. Dhaouadi, F. Touati and N. Gharbi, Nano-Micro Lett., 2011, 3, 242–248 CrossRef CAS.
- K. G. Bhattacharyya and A. K. Talukdar, Catalysis in Petroleum and Petrochemical Industries, Narosa Publishing House, 2005, p. 272 Search PubMed.
- W. Smith, A. Wolcott, R. C. Fitzmorris, J. Z. Zhang and Y. Zhao, J. Mater. Chem., 2011, 21, 10792–10800 RSC.
- J. Gierer, E. Yang and T. Reitberger, Holzforschung, 1994, 48, 405–413 CrossRef CAS.
- O. Karlsson, T. Ikeda, T. Kishimoto, K. Magara and Y. Matsumoto, International symposium of wood and pulp conference, Yokohama, 1999, vol. 1, pp. 178–181 Search PubMed.
- O. Karlsson, T. Ikeda, T. K. K. Magara, Y. Matsumoto and S. Hosoya, J. Wood Sci., 2000, 46, 263–265 CrossRef CAS.
- T. Akiyama, T. Sugimoto, Y. Matsumoto and G. Meshitsuka, J. Wood Sci., 2002, 48, 210–215 CrossRef CAS.
- D. Shao, J. Liang, X. Cui, H. Xu and W. Yan, Chem. Eng. J., 2014, 244, 288–295 CrossRef CAS.
- Y. Hamzeh, G. Mortha, D. Lachenal, J. C. Hostachy and C. Calais, J. Wood Chem. Technol., 2006, 2, 153–164 CrossRef.
- T. Furusawaa, T. Satoa, M. Saitoa, Y. Ishiyamaa, M. Satob, N. Itoha and N. Suzuki, Appl. Catal., A, 2007, 327, 300–310 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |