
Continuous supercritical route for quantum-confined GaN nanoparticles†
B.
Giroire
ab,
S.
Marre
ab,
A.
Garcia
ab,
T.
Cardinal
ab and
C.
Aymonier
*ab
aCNRS, ICMCB, UPR 9048, F-33600 Pessac, France. E-mail: cyril.aymonier@icmcb.cnrs.fr
bUniv. Bordeaux, ICMCB, UPR 9048, F-33600 Pessac, France
First published on 4th November 2015
Abstract
GaN quantum dots (QDs) are prepared in a one-step continuous process using anhydrous solvents at supercritical conditions (and temperatures below 450 °C) in short residence times, typically less than 25 s. The as-prepared QDs exhibit a strong luminescence in the ultraviolet (λmax ~ 350 nm) in agreement with the synthesis of quantum-confined GaN nanoparticles.
Gallium nitride (GaN) gained increasing interest over the past twenty years thanks to its excellent optoelectronic properties, high thermal stability and biocompatibility.1,2 GaN quantum dots generally present a zinc blend structure, exhibiting a size-dependent bandgap when the particle radius is below the Bohr exciton radius, i.e. 2.5 nm for GaN.3 This III–V nitride direct bandgap semiconductor has been mainly used for the preparation of UV-to-blue LEDs.4,5 Recently, GaN QDs have been tested in photovoltaic cells,6 gas sensors,7 and as photocatalysts.8,9 However, these applications require enhanced control over the synthesis processes and the development of powerful and efficient synthetic techniques. Indeed, it was underlined in a recent review paper of Fan et al. that the quality of GaN nanomaterials prepared via solution routes is still too low for the aforementioned applications.10 Thus, the chemical synthesis of colloidal GaN nanoparticles remains challenging.
A wide range of synthesis reactions have been reported. The involved precursors include gallium halides,11 azides,12,13 and nitrogen-rich molecular precursors such as cyclotrigallazane,14 polymeric gallium imide,15 as well as tris(dimethylamido)gallium(III) ([Ga(NMe2)3]2).16 These methods require temperatures above 250 °C, long reaction times (>24 h) and control of the reaction atmosphere to yield crystalline and oxygen-carbon-free gallium nitride.17 More recently, a promising ammonia-in-oil micro-emulsion synthetic pathway at low temperature (T = −40 °C) was developed leading to UV-emitting GaN QDs.18 However, all these synthesis approaches were performed in batch mode, which can suffer from lack in reproducibility and limited control of the operating conditions.
Continuous microfluidics19,20 and supercritical fluids routes have been demonstrated to be efficient approaches, for the synthesis of mainly oxides,21,22 semiconductors,23,24 and metals nanoparticles,25 with control over size, morphology, crystallinity and composition. Only one study reported the synthesis of copper, nickel and cobalt nitrides in supercritical ammonia from acetylacetonate precursors.26 Nevertheless, this method applied to gallium – exhibiting a higher affinity with oxygen – only yielded pure gallium oxide.
In this study, we report the first fast and continuous synthesis of GaN QDs in a tubular microreactor at supercritical conditions. Our approach combines both the advantages of supercritical fluids and microscale reactors,27 leading to a precise control of the reaction environment and the experimental conditions. This synthesis involves the decomposition of the tris(dimethylamido)gallium(III) dimer in supercritical cyclohexane or hexane–ammonia mixture.
In a typical experiment, the precursor solution is injected via the inner flow as shown in Fig. 1. Pure solvent is injected through the outer flow (with possible addition of surfactant). The co-flow geometry confines nucleation at the center of the tubing, i.e. far from the reactor walls, thus preventing deposition and clogging of the setup. The two solutions are injected using two high pressure pumps operating at 150 bar. The nanoparticles were first prepared in anhydrous cyclohexane (Tc = 281 °C, pc = 40.7 bar) at 400 °C (process 1, Fig. 1). In such conditions, the residence time is determined to be 22 seconds (calculation available in ESI†). Different experiments were also performed using ammonia (Tc = 132.3 °C, pc = 113.3 bar) as the co-solvent. In these experiments, [Ga(NMe2)3]2 is injected in the reactor in a hexane solution. The ammonia–hexane mixture was brought to the system at 350 °C and 160 bar (process 2, Fig. 1). Additional details for sample preparation and characterization are available in the footnotes.‡§
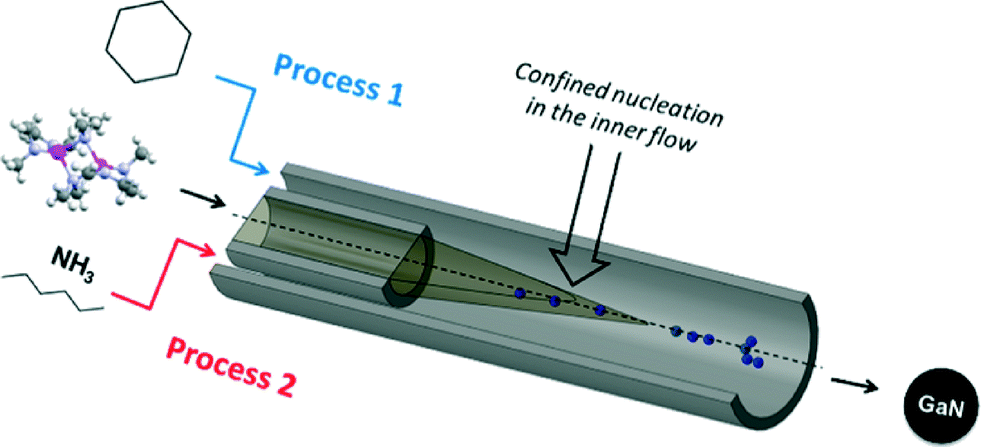 | ||
| Fig. 1 Schematic representation of the mixing zone of the coflow setup. | ||
The size and morphology of the particles obtained were investigated by Transmission Electron Microscopy (TEM). Representative pictures are shown in Fig. 2a and b. Nanoparticles of 3.1 nm in diameter were measured. Lattice fringes can be identified in high resolution pictures, available in Fig. S1 (see ESI†). A d-spacing of 0.26 nm was measured corresponding to the <111> plane of cubic GaN. Partial aggregation was observed due to high particle surface energy and lack of surface stabilizer in the reaction media. Structure of the as-prepared material in supercritical fluids was determined by powder X-ray diffraction. GaN can exhibit different crystalline phases: cubic (zinc blende) and hexagonal (würtzite) at ambient conditions, displaying bandgaps of 3.3 eV and 3.5 eV, respectively.28 An additional metastable cubic phase (rock-salt) can also be detected at high pressure.29,30 The XRD diagram obtained is shown in Fig. 2c. Synthesis from the single source precursor in supercritical cyclohexane yields material exhibiting two broad diffraction bands, in agreement with nanosized GaN presenting a cubic phase described elsewhere (inset Fig. 2c).16,18
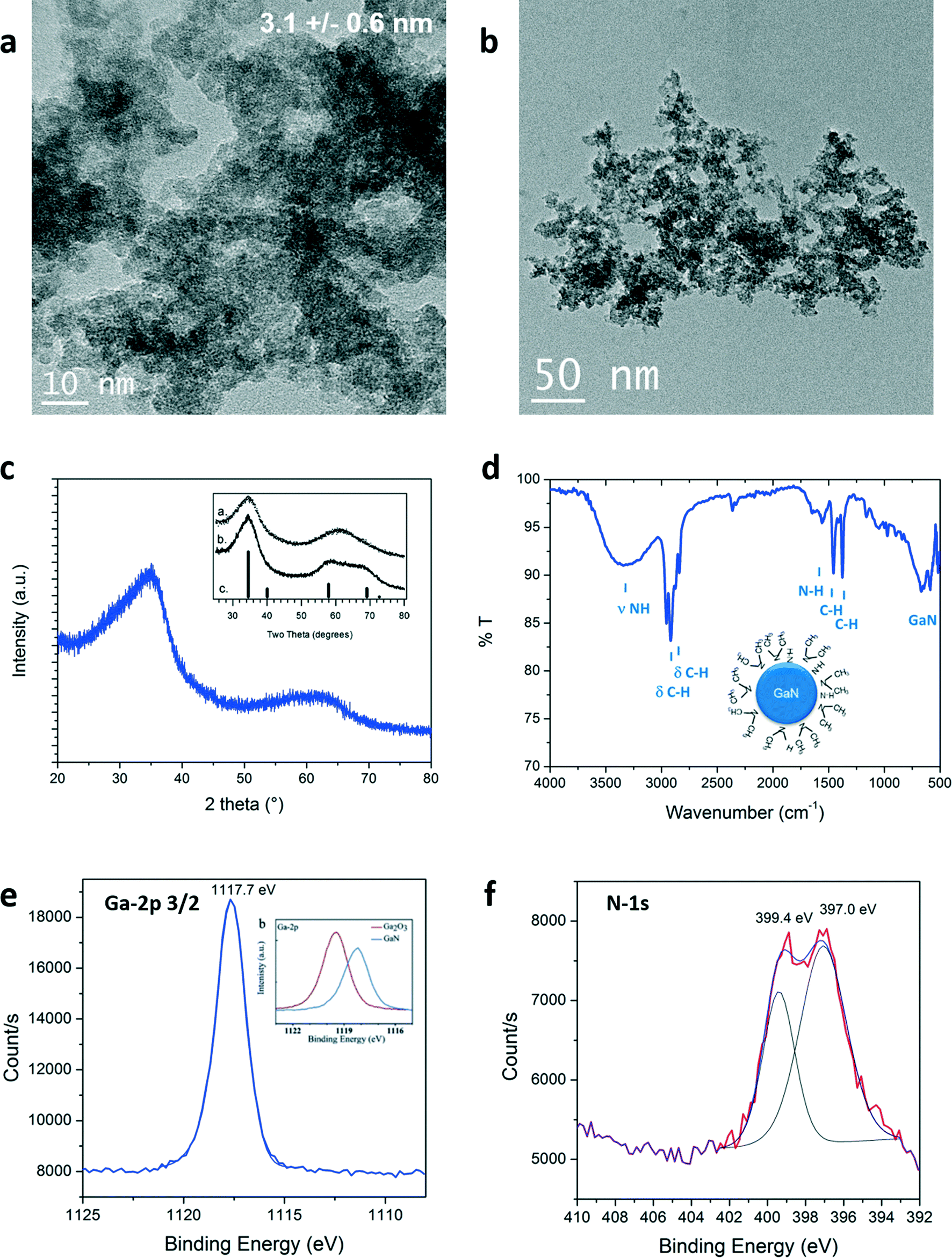 | ||
| Fig. 2 a), b) TEM images, c) XRD pattern (with inset from ref. 16), d) ATR-FTIR, e) and f) Ga-2p3/2 (with inset from ref. 31) and N-1s XPS signals of the GaN quantum dots prepared in supercritical cyclohexane at 400 °C and 150 bar. | ||
Nanoparticles of such sizes are expected to display quantum confinement effect. Note that it is possible to obtain stable solutions by post-synthesis treatments such as high power sonication (probe 400 W), as well as ex situ or in situ surface modification.
The quality of the as-prepared materials was checked by X-ray photoelectron spectroscopy (XPS). Signals corresponding to Ga, N, C and O were detected. The complete survey as well as C-1s and O-1s signals are available in the ESI.† The carbon and oxygen signatures could partially be attributed to surface contamination, inherent to our sample preparation for this characterization technique (air contact) and/or to the carbon tape holder. XPS spectra of Ga-2p3/2 and N-1s are displayed in Fig. 2e and f, respectively. The presence of the Ga-2p3/2 signal at 1117.7 eV confirmed that gallium nitride was formed.31 Gallium oxide would be expected to exhibit higher binding energies (see onset of Fig. 2e, Ga-2p3/2: 1119.5 eV).31,32 Moreover, two contributions could be fitted to the N-1s spectra: one centered at 397.0 eV, in agreement with the formation of nitride bonding,31 the other, at 399.4 eV corresponds to the binding of nitrogen to a sp3 carbon.33 The same contribution was observed in the C-1s signal at 286.1 eV (see ESI†). This signal could be arising from the presence of –N(CH3)2 groups. This assumption could be clarified by surface investigation. Indeed, the ATR-FTIR analysis, seen in Fig. 2d, showed bands confirming the presence of N–H but also –N(CH3)2 groups that should be present at the particle surface. The presence of this specific surface species is attributed to in situ functionalization of the nanoparticles by trimethylamine or dimethylamine by-products formed during the decomposition of the precursor. The XPS analysis clearly demonstrates the successful synthesis of GaN nanostructures. Furthermore, the lack of contribution at lower binding energies, below 1116 eV in the Ga-2p as well as below 18 eV in the Ga-3d spectra (available in ESI†), proves the synthesis of metallic gallium-free material. Note that it was also possible to identify cubic GaN in syntheses from [Ga(NMe2)3]2 prepared at temperature as low as 300 °C and pressure as low as 69 bar (see ESI†).
In similar experiments, ammonia could be added in the system via the outer flow (process 2, Fig. 1). The single source precursor was injected via the inner flow in a hexane solution (using the same concentration). Hexane (Tm = −95 °C) was used instead of cyclohexane (Tm = 6.5 °C) to avoid freezing of the solution at the system outlet. The mixture was brought to 350 °C and 160 bar and the total flowrate was fixed at 1 mL min−1, resulting in a residence time of approximately 14 seconds (calculation available in ESI†). Materials prepared by this route also exhibit a cubic phase. A particle size of approximately 2.8 nm could be measured for this sample, as shown in the TEM picture displayed in Fig. 3a. Generally, the particles prepared with and without ammonia presented similar characteristics. However, different surface functionalities could be identified, as shown in Fig. 3b. Indeed, the surface of the GaN nanoparticles prepared in ammonia only displayed surface N–H bonds. This different behaviour can be explained by a difference in the reaction mechanism. Janik et al. demonstrated that [Ga(NMe2)3]2 readily form polymeric gallium imide {Ga(NH)3/2}n in the presence of liquid or gaseous ammonia at room temperature.34 In our experiment, liquid ammonia and [Ga(NMe2)3]2 encountered at the mixing point at RT, forming the polymeric gallium imide that condenses and nucleates to GaN at elevated temperature. When ammonia was not present in the reaction medium, only thermal decomposition/condensation (possibly through a radical mechanism) of [Ga(NMe2)3]2 could occur,35 leaving unreacted dimethylamine groups at the nanoparticles surface. Also, the presence of these groups could arise from the dimethylamine/trymethylamine by-products present in solution near the nuclei.
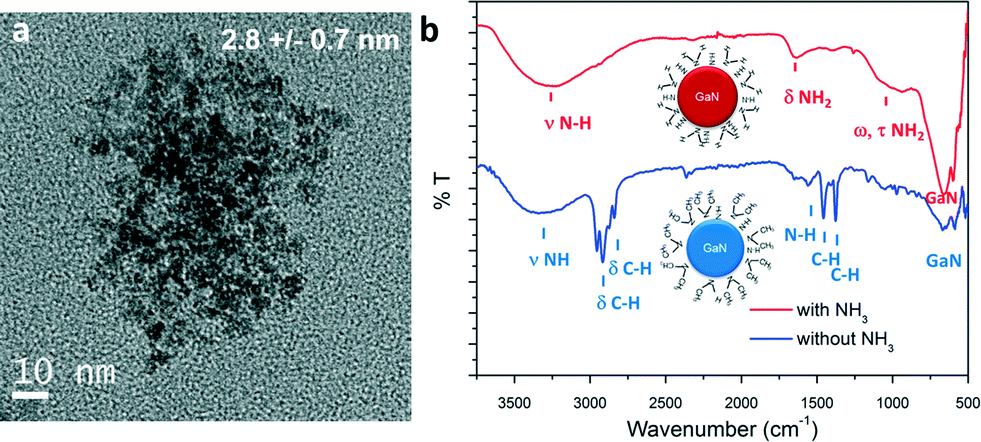 | ||
| Fig. 3 a) TEM image and b) ATR-FTIR spectra of GaN QDs synthesized with and without ammonia, schemes of particles with surface functionalities are available in inset. | ||
Optical characterizations were performed on the GaN QDs in solutions. High intensity room temperature photoluminescence (PL) of the nanoparticles in solution was recorded in the UV region, as seen in Fig. 4. The dotted lines correspond to the excitation spectra and the plain lines represent the emission spectra of the as-prepared GaN nanoparticles. The blue and red curves correspond to the reaction performed in cyclohexane only and in the presence of ammonia, respectively. UV, near band-edge emissions were measured at room temperature on the as-prepared samples with λmax = 343 nm and λmax = 335 nm. The recorded emission peak displayed a FWHM of 0.5 eV, narrower than what is observed in the literature (usually >0.65 eV) implying a narrow size distribution of the emitting centers. Typically, microscale hexagonal and cubic GaN exhibits an emission closer to the 370–400 nm range.36–38 The blue-shift of both near-bandgap emissions displays evidence of quantum-confinement. Looking at the excitation curve, second features are observed at 255 nm (NPs prepared in cyclohexane) and 228 nm (NPs prepared with ammonia). Upon excitation at these wavelengths, emissions centered at the same maximum wavelengths were measured. It was concluded that this second feature corresponds to the second energy levels of quantum confined GaN, due to discretization of the energy levels in the band structure of the material.39 Furthermore, no emission signals in the visible were recorded in our samples, implying the formation of defect-free GaN. The slight increase of the FWHM of approximately 0.1 eV of the emission band for the sample prepared with ammonia is in agreement with a broader nanoparticle size distribution, as shown in Fig. 2 and 3.
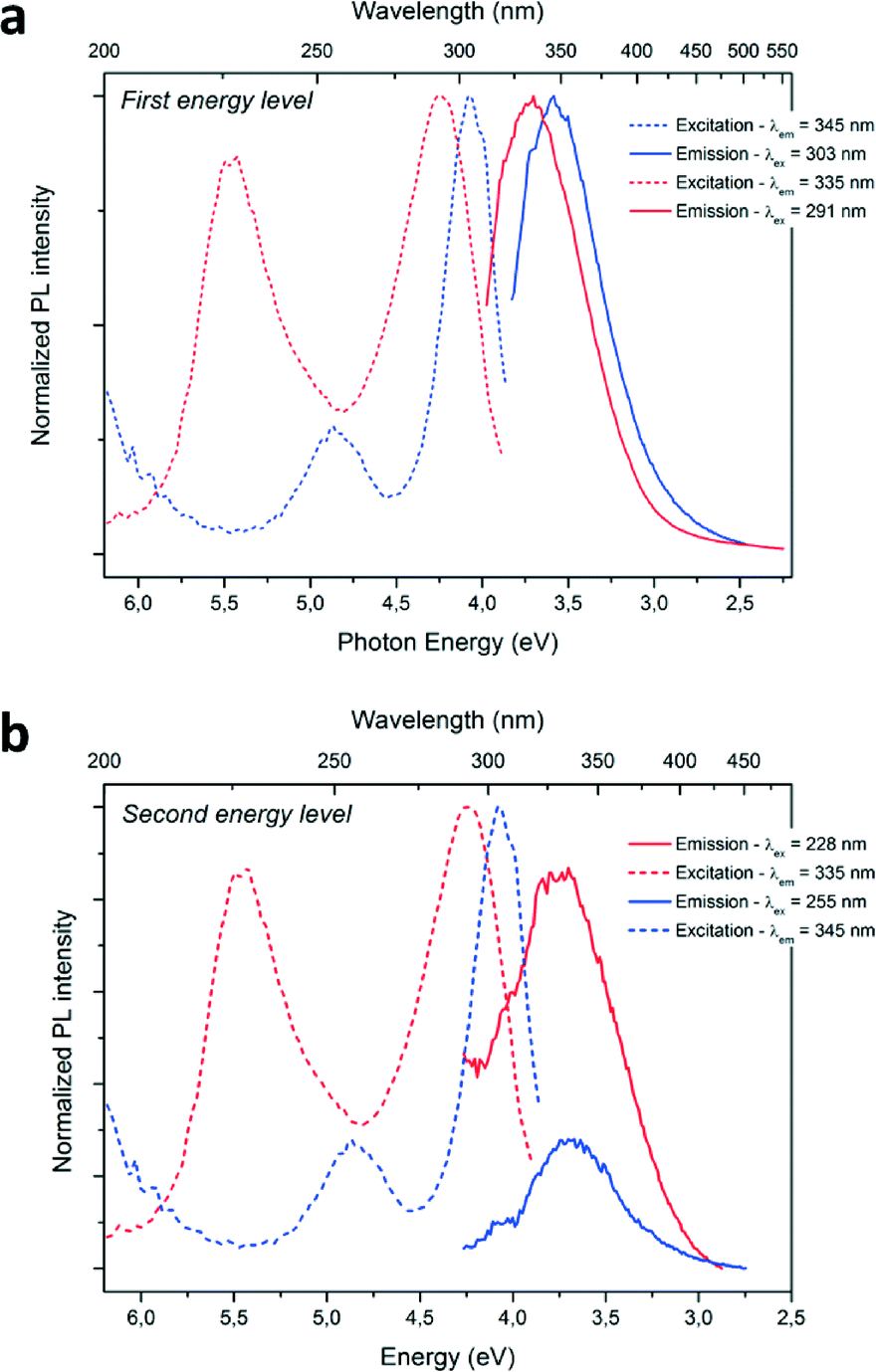 | ||
| Fig. 4 a) b) Photoluminescence spectra of the sample prepared: in cyclohexane are represented in blue, in the ammonia–hexane mixture are represented in red; with excitation spectra in dotted line and emission spectra in plain line. | ||
A concurrent shift of both emission (from λmax = 345 nm to λmax = 335 nm; ΔE = 0.14 eV) and excitation (from λmax = 303 nm to λmax = 291 nm; ΔE = 0.19 eV) maximum wavelengths towards the UV are observed for the PL spectra when ammonia was added to the reaction media. This change can be explained by a difference in particle size. Indeed, a diminution of the average size of the particles from 3.1 to 2.8 nm is measured for the particles prepared with ammonia. The PL maxima shifts with particle size are therefore an evidence of size-related properties. The energy difference between the two main excitation features increases from 0.78 eV to 1.23 eV, when the particles size decreases from 3.1 to 2.8 nm and could be due to different band structure induced by the difference in particle size. Further discretization of the energy levels is expected when decreasing particle size for GaN QDs, similarly to previous studies concerning II–VI QDs (observable in absorption spectra of the following references).39–41
UV-visible absorption spectrum presents blue-shifted onsets of absorption at 3.95 eV (process 1) and 4.08 eV (process 2) (see ESI,† Fig. S5a). Optical bandgaps of 4.21 and 4.52 eV for GaN NPs prepared in processes 1 and 2, respectively, were determined with a Tauc plot (see ESI,† Fig. S5b). The important increase of the bandgap (>0.9 eV) compared to bulk cubic-GaN (3.3 eV) correlated with the small size of the nanoparticles measured by TEM and the blue shifted photoluminescence emissions confirms the formation of quantum-confined GaN.
In summary, we have demonstrated the first one-step, fast and continuous synthesis of gallium nitride QDs at supercritical conditions. The presented setup allows a fine control of the experimental conditions resulting in high homogeneity of the reaction medium. Cubic gallium nitride nanoparticles of 3.1 nm diameter were identified. Optical characterization of the as-prepared materials shows evidences of quantum confinement. A blue shift of the bandgap of more than 0.9 eV could be determined from UV spectroscopy. A high intensity, UV, near band-edge emission was detected (λmax = 343 nm) as well as a second excitation level emission confirming the formation of quantum-confined GaN. The narrow PL FWHM as well as the XPS spectra confirm the synthesis of high quality material. The addition of ammonia yielded cubic material with different surface functionalities, which was attributed to a change in the reaction mechanism. Additionally, the reproducible continuous synthesis approach could be scaled up, allowing realizing the promise of GaN QDs in applications such as LEDs devices, gas sensing and photocatalysis.
Acknowledgements
The authors would like to acknowledge the “Agence Nationale de la Recherche” (convention no. ANR-12-G8ME-0002-01), and the region Aquitaine for their financial support.Notes and references
- F. A. Ponce and D. P. Bour, Nature, 1997, 386, 351–359, DOI:10.1038/386351a0 , Publ. Online 27 March 1997.
- M. Razeghi and A. Rogalski, J. Appl. Phys., 1996, 79, 7433–7473 CrossRef CAS.
- H. Morkoç, S. Strite, G. B. Gao, M. E. Lin, B. Sverdlov and M. Burns, J. Appl. Phys., 1994, 76, 1363–1398 CrossRef.
- S. Nakamura, T. Mukai and M. Senoh, Jpn. J. Appl. Phys., 1991, 30, L1998 CrossRef CAS.
- K. Iso, H. Yamada, H. Hirasawa, N. Fellows, M. Saito, K. Fujito, S. P. DenBaars, J. S. Speck and S. Nakamura, Jpn. J. Appl. Phys., 2007, 46, L960 CrossRef CAS.
- D. V. P. McLaughlin and J. M. Pearce, Metall. Mater. Trans. A, 2013, 44, 1947–1954 CrossRef CAS.
- B. Chitara, D. J. Late, S. B. Krupanidhi and C. N. R. Rao, Solid State Commun., 2010, 150, 2053–2056 CrossRef CAS.
- H. S. Jung, Y. J. Hong, Y. Li, J. Cho, Y.-J. Kim and G.-C. Yi, ACS Nano, 2008, 2, 637–642 CrossRef CAS PubMed.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- G. Fan, C. Wang and J. Fang, Nano Today, 2014, 9, 69–84 CrossRef CAS.
- Y. Xie, Y. Qian, W. Wang, S. Zhang and Y. Zhang, Science, 1996, 272, 1926–1927 CAS.
- J. Choi and E. G. Gillan, J. Mater. Chem., 2006, 16, 3774 RSC.
- A. Manz, A. Birkner, M. Kolbe and R. A. Fischer, Solution Synthesis of Colloidal Gallium Nitride at Unprecedented Low Temperatures, Adv. Mater., 2000, 12, 569–573 CrossRef CAS.
- R. J. Jouet, A. P. Purdy, R. L. Wells and J. F. Janik, J. Cluster Sci., 2002, 13, 469–486 CrossRef CAS.
- O. I. Mićić, S. P. Ahrenkiel, D. Bertram and A. J. Nozik, Appl. Phys. Lett., 1999, 75, 478 CrossRef.
- G. Pan, M. E. Kordesch and P. G. Van Patten, Chem. Mater., 2006, 18, 3915–3917 CrossRef CAS.
- M. A. Reshchikov and H. Morkoç, J. Appl. Phys., 2005, 97, 061301 CrossRef.
- F. Gyger, P. Bockstaller, H. Gröger, D. Gerthsen and C. Feldmann, Chem. Commun., 2014, 50, 2939 RSC.
- S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183–1202 RSC.
- E. Shahbazali, V. Hessel, T. Noël and Q. Wang, Nanotechnol. Rev., 2013, 3, 65–86 Search PubMed.
- F. Cansell and C. Aymonier, J. Supercrit. Fluids, 2009, 47, 508–516 CrossRef CAS.
- Y. Roig, S. Marre, T. Cardinal and C. Aymonier, Angew. Chem., Int. Ed., 2011, 50, 12071–12074 CrossRef CAS PubMed.
- S. Marre, J. Park, J. Rempel, J. Guan, M. G. Bawendi and K. F. Jensen, Adv. Mater., 2008, 20, 4830–4834 CrossRef CAS.
- A. Chakrabarty, S. Marre, R. F. Landis, V. M. Rotello, U. Maitra, A. D. Guerzo and C. Aymonier, J. Mater. Chem. C, 2015, 3, 7561–7566 RSC.
- T. Gendrineau, S. Marre, M. Vaultier, M. Pucheault and C. Aymonier, Angew. Chem., Int. Ed., 2012, 51, 8525–8528 CrossRef CAS PubMed.
- S. Desmoulins-Krawiec, C. Aymonier, A. Loppinet-Serani, F. Weill, S. Gorsse, J. Etourneau and F. Cansell, J. Mater. Chem., 2004, 14, 228–232 RSC.
- S. Marre, Y. Roig and C. Aymonier, J. Supercrit. Fluids, 2012, 66, 251–264 CrossRef CAS.
- S. Chichibu, H. Okumura, S. Nakamura, G. Feuillet, T. Azuhata, T. Sota and S. Yoshida, Jpn. J. Appl. Phys., 1997, 36, 1976 CrossRef CAS.
- A. Muoz and K. Kunc, Phys. Rev. B, 1991, 44, 10372 CrossRef.
- H. Xia, Q. Xia and A. L. Ruoff, Phys. Rev. B, 1993, 47, 12925 CrossRef CAS.
- S. Bhaviripudi, J. Qi, E. L. Hu and A. M. Belcher, Nano Lett., 2007, 7, 3512–3517 CrossRef CAS PubMed.
- M. Kumar, S. K. Pasha, T. C. Shibin Krishna, A. P. Singh, P. Kumar, B. K. Gupta and G. Gupta, Dalton Trans., 2014, 43, 11855 RSC.
- S. E. Rodil, N. A. Morrison, J. Robertson and W. I. Milne, Phys. Status Solidi, 1999, 174, 25–37 CrossRef CAS.
- J. F. Janik and R. L. Wells, Chem. Mater., 1996, 8, 2708–2711 CrossRef CAS.
- A. L. Hector, Chem. Soc. Rev., 2007, 36, 1745 RSC.
- D. Esken, S. Turner, C. Wiktor, S. B. Kalidindi, G. Van Tendeloo and R. A. Fischer, J. Am. Chem. Soc., 2011, 133, 16370–16373 CrossRef CAS PubMed.
- T. Kuykendall, P. Ulrich, S. Aloni and P. Yang, Nat. Mater., 2007, 6, 951–956 CrossRef CAS PubMed.
- Y. Hori, X. Biquard, E. Monroy, D. Jalabert, F. Enjalbert, L. S. Dang, M. Tanaka, O. Oda and B. Daudin, Appl. Phys. Lett., 2004, 84, 206 CrossRef CAS.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS PubMed.
- S. M. Reimann and M. Manninen, Rev. Mod. Phys., 2002, 74, 1283 CrossRef CAS.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5re00039d |
‡ Experimental setup and material synthesis: tris(dimethylamido)gallium(III) ([Ga(NMe2)3]2, Sigma-Aldrich, 98%) was dissolved in high purity, anhydrous alkane solutions: cyclohexane (99.5%, Sigma-Aldrich) or hexane (≥99%, Sigma-Aldrich). Concentration of the precursor in the reactor was fixed at 5 × 10−3 mol L−1. The solution preparation was performed under controlled atmosphere to prevent water contamination. All chemicals were handled without further purification. Solutions were injected into the reactor using Jasco PU 2080 plus high pressure pumps. The reactor consists of a two-meter long 1/16′′ 316 L stainless steel tubing coiled around a heating cartridge (Acim Jouanin, 800 W, 230 V) controlling the temperature up to 450 °C. The internal diameter of the tubing was 1 mm, corresponding to a volume of 1.60 mL. An adjustable back pressure regulator (Idex-hs, P-880) allowed a control of the pressure up to 345 bar. A concurrent flow regime was established inserting an inner 316 L stainless steel tubing (OD: 1/32′′, ID: 400 μm) inside the main tubing at the entrance of the reactor. The gallium precursor was pumped through the inner tubing. The total flow rate was fixed at 2 mL min−1. Ammonia (NH3 100%, Air Liquide, Tc = 132.3 °C, pc = 113.3 bar) was liquefied in an ISCO 100DM syringe pump and pressurized before being injected in the system via the outer flow. [Ga(NMe2)3]2 was injected in hexane (Tc = 234.7 °C, pc = 30.3 bar) via the inner flow. The ammonia to hexane ratio was fixed at 1 to 1 vol (corresponding to a hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ammonia molar ratio of 0.17:0.83). The critical coordinates of this mixture could be determined from Refprop®: Tc = 138,6 °C, pc = 92.5 bar. Reactions with ammonia and hexane were carried out at 350 °C and 150 bar. The total flow rate was fixed at 1 mL min−1. Powder sample could be collected after centrifugation and washing of the solution. :ammonia molar ratio of 0.17:0.83). The critical coordinates of this mixture could be determined from Refprop®: Tc = 138,6 °C, pc = 92.5 bar. Reactions with ammonia and hexane were carried out at 350 °C and 150 bar. The total flow rate was fixed at 1 mL min−1. Powder sample could be collected after centrifugation and washing of the solution. |
| § Characterization techniques: X-ray diffraction (XRD) patterns were collected on a PANalitycal X'pert MPD-PRO Bragg–Brentano θ–θ geometry diffractometer equipped with a secondary monochromator over an angular range of 2θ = 8–80°. The Cu-Kα (λ = 0.15418 nm) radiation was generated at 45 kV and 40 mA. High resolution electron microscopy (HRTEM) was performed using a Jeol 2200FS, working at 200 kV. Samples were prepared by deposition of diluted nanoparticles dispersion solutions onto carbon-copper grids. Optical characterizations were carried out in liquid suspension. Luminescence spectra were recorded on a Fluorolog FL3 22 iHR320 spectrofluorimeter (Horiba Jobin Yvon) with two double grating monochromators (excitation and emission for the UV-vis studies). The excitation source is a 450 Watt xenon lamp, excitation spectra were corrected for the variation of the incident lamp flux, as well as emission spectra for the transmission of the monochromator and the response of the photomultiplier (R928P photomultiplier). UV-vis absorption spectra were acquired using a Carry 5000 (Agilent Technologies) in the 200–800 nm range. ATR-FTIR was carried out on solid samples using a MIRacle 10 from Shimadzu. XPS spectra were recorded on a Thermo Scientific K-Alpha spectrometer; a spot size of 250 μm was employed. |
| This journal is © The Royal Society of Chemistry 2016 |