
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor–acceptor single cocrystal of coronene and perylene diimide: molecular self-assembly and charge-transfer photoluminescence†
Chen Wanga,
Jianlin Wangb,
Na Wua,
Miao Xua,
Xiaomei Yanga,
Yalin Lubc and
Ling Zang*a
aNano Institute of Utah, Department of Materials Science and Engineering, University of Utah, Salt Lake City, UT 84112, USA. E-mail: lzang@eng.utah.edu
bNational Synchrotron Radiation Laboratory, Synergetic Innovation Center of Quantum Information & Quantum Physics, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
cLaser Optics Research Center, Department of Physics, United States Air Force Academy, CO 80840, USA
First published on 28th November 2016
Abstract
The research on donor–acceptor (D–A) cocrystalline structures based on organic semiconductive molecules has drawn great attention due to their unique optical and electronic properties. Among the building block molecules, derivatives of perylene-3,4,9,10-tetracarboxylic-3,4,9,10-diimides (PTCDI) are of particular interest as these molecules form high performance n-type semiconductors with strong air stability. However, the cocrystal of PTCDIs remains challenging to fabricate, and only few D–A cocrystals of PTCDIs have been reported. Herein, we report a successful molecular self-assembly design for the PTCDI cocrystal with a donor molecule, coronene. Within the triclinic cell of the cocrystal, the PTCDI and coronene molecules achieved 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 alternated π–π stacking. The cocrystal showed clear red-shifted absorption and photoluminescence bands, implying the strong charge transfer interaction between coronene and PTCDI. Additionally, the cofacial π–π stacking between coronene and PTCDI planes favors strong one-dimensional self-assembly, leading to the formation of microsized fibril cocrystals and arrays. This presented cocrystal design strategy helps to explore new D–A cocrystalline structures, particularly with one-dimensional morphology control.
:1 alternated π–π stacking. The cocrystal showed clear red-shifted absorption and photoluminescence bands, implying the strong charge transfer interaction between coronene and PTCDI. Additionally, the cofacial π–π stacking between coronene and PTCDI planes favors strong one-dimensional self-assembly, leading to the formation of microsized fibril cocrystals and arrays. This presented cocrystal design strategy helps to explore new D–A cocrystalline structures, particularly with one-dimensional morphology control.
Introduction
The charge transfer (CT) interaction in donor–acceptor (D–A) materials and the relevant molecular self-assembly (or microstructure) have drawn increasing attention in recent years.1,2 The D–A cocrystal structures, in a format of homogeneous assembly of the D–A molecules, may possess unique optical or electrical properties, such as high conductivity,3 ambipolar carrier transport,4,5 ferroelectricity,6 and tunable luminescence,7 which enable many applications in electronic or optoelectronics devices. However, the design and growth of D–A cocrystals are still challenging, especially for molecules with low solubility,8 since these molecules are strongly in favor of self-aggregation into D or A assemblies alone, rather than D–A cocrystals. Among the building block molecules studied for organic semiconductors, molecules of perylene-3,4,9,10-tetracarboxylic-3,4,9,10-diimides (PTCDI, Scheme 1) represent one of the most interesting classes for several unique features correlated real applications in devices.9–11 First, PTCDIs form a rare class of air-stable n-type organic semiconductors, and only few of such n-type materials have been employed in optoelectronic devices (e.g., photovoltaics, LEDs, sensors). Second, the two imide positions in PTCDI are nodes in the front π-orbitals, and thereby the side-chain modification at the two ends does not affect the electronic property of PTCDI, offering unlimited options for structural modification to tune and optimize the π–π stacking conformation. Third, the band gap of PTCDI is about 2.5 eV, implying strong absorption in the visible region, which is desired for development into solar energy utilization. Lastly, the rigid, planar π-conjugation of PTCDI structure makes it suited for cofacial molecular stacking, which usually leads to formation of one-dimensional fiber like materials with uniaxial optical and electrical properties.12,13 But also due to the strong π–π self-stacking, PTCDI molecules intend to form homogeneous assembly of themselves, even in the presence of electron donor molecules.14–19 To form D–A cocrystals, the molecular structure must be designed so that the D–A interaction exceeds the π–π stacking between PTCDIs, while still maintains the overall geometry matching for the cofacial intermolecular arrangement for a crystalline phase. Because of this structural design challenge, few D–A cocrystal structures of PTCDI was reported,5 and the high quality cocrystals are thereby highly desired. The single crystalline data thus obtained will provide the theoretical calculation and modeling with precise geometry and configuration of intermolecular arrangement that are crucial for studying the structure–property relationship. | ||
| Scheme 1 Molecular structures of coronene and PTCDI-C6. | ||
The donor molecule used in this study was coronene (Scheme 1), which possesses a rigid and planar π-conjugation, and is correlated to the core of PTCDI (regarding size and geometry). The matching π–π stacking between coronene and PTCDI, in addition to the D–A interaction, is expected to overcome the self-stacking of PTCDIs, leading to the formation of D–A cocrystal as indeed confirmed later in this work. Our X-ray diffraction (XRD) study proved the single crystal phase of the D–A assembly of coronene–PTCDI, in which the coronene and PTCDI molecules stack 1:1 alternately in a triclinic crystal cell structure. The significant D–A interaction can be revealed from the CT absorption band in longer wavelength measured over the cocrystal, which becomes in dark purple color in comparison to the light yellow coronene and red color PTCDI crystals. The cofacial π–π stacking of D–A molecules, in cooperation with the moderate hydrogen bonding between adjacent PTCDIs, facilitates the one-dimensional (1D) growth of the molecular self-assembly, leading to formation of fibril cocrystals in micrometer size and further fabrication of microfiber array. And beyond the structure, this stacking feature could further results in the clear uniaxial optical property of these 1D microstructures, as shown by the highly polarized photoluminescence.
Results and discussion
Coronene can be molecularly dissolved in chloroform, and the UV-vis absorption and fluorescence spectra of the solution show highly structured peaks in the region 250–350 and 400–500 nm, respectively (Fig. 1a). The PTCDI molecule selected in this study is the one substituted with two cyclohexyl groups (–C6), namely PTCDI-C6 (Scheme 1). PTCDI-C6 can also be dissolved in chloroform, forming homogeneous molecular solution, which demonstrates the typical absorption and fluorescence (photoluminescence) spectra of PTCDI shown in Fig. 1a. In solid state, the absorption and fluorescence of coronene is red-shifted (compared to the molecular solution) due to the molecular aggregation, consistent with the light yellow color in appearance and brightly green photoluminescence.20,21 Solid state of PTCDI-C6 shows broad absorption ranging up to 640 nm and excimer emission centered at 650 nm (Fig. 1b, c and S1†). In comparison to the pure solid of coronene and PTCDI, the cocrystal powders appears in dark purple color (Fig. S1a†), which is consistent with the new absorption band in longer wavelength region from 650 nm to 700 nm (Fig. 1b). Such significant red-shifted absorption band is usually referred as the donor–acceptor CT band, which was also observed in other donor–acceptor systems containing PTCDI structures.22,23 Correspondingly, the photoluminescence of the cocrystal is also red-shifted to 730 nm (with quantum yield ΦPL = 5.5%). To confirm that the photoluminescence of the cocrystal was generated from the CT transition, the excitation spectra of solid PTCDI-C6 and the cocrystal were measured and compared (Fig. S2†). The result showed that the luminescence of cocrystal is mostly contributed by the new D–A absorption band, whereas the relative luminescence of PTICD-C6 at different wavelengths matches well the absorption spectra (Fig. 1b). Additionally, such CT luminescence was found to be temperature-dependent (Fig. 1d). At 77 K, the luminescence peak was blue-shifted to 660 nm and became asymmetric, and meanwhile the intensity was increased by more than five times. The increased emission is ascribed to the weakened nonradiative decay, which is likely caused by the more localized frontier molecular orbitals.4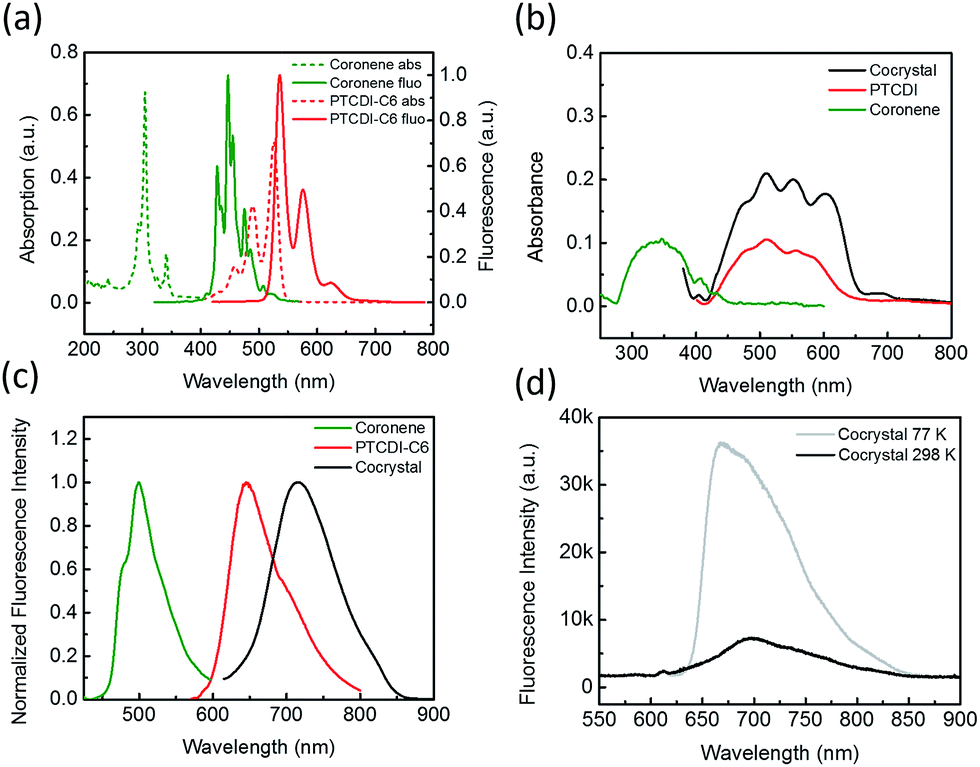 | ||
| Fig. 1 (a) Absorption (dash lines) and fluorescence (solid lines) spectra of chloroform solution of coronene (green, 1 mmol L−1) and PTCDI-C6 (red, 1 mmol L−1). (b) Absorption spectra (baseline corrected) measured on the solid sample of coronene (green), PTCDI-C6 (red) and the cocrystal (black). (c) Fluorescence spectra measured on the solid sample of coronene (green), PTCDI-C6 (red) and the cocrystal (black). (d) Fluorescence spectra of the cocrystal measured at room temperature (298 K, black) and 77 K (gray) on the same area of the sample and under the same photoexcitation intensity. | ||
With the single crystal structure, we performed the density-functional theory (DFT) calculation for the molecular orbitals based on a D–A stack model, for which the two cyclohexyl side groups were replaced with hydrogen to simplify the calculation. Because the two nitrogen atoms of the diimide of PTCDI are nodes in π-orbitals, changing the side group does not significantly change the electronic property of PTCDI. The calculated lowest unoccupied molecular orbitals (LUMO), the highest occupied orbitals (HOMO), and the next two lower orbitals HOMO-1 and HOMO-2 are shown in Fig. S3.† And the results clearly demonstrated that the LUMO of the cocrystal only locates in the PTCDI molecules. So under light irradiation, the CT process may occur from the coronene to PTCDI-C6, as indicated by the electron transition from HOMO, HOMO-1 and HOMO-2 to LUMO, which results in electron redistribution and localization in PTCDI part (Table S1†). These calculations support well the photoinduced CT observed by the spectral measurements (Fig. 1).
XRD measurement of coronene–PTCDI cocrystal helped to resolve the crystalline structure as shown in Fig. 2a. Within the cocrystal, the coronene and PTCDI-C6 molecules stack alternately along the a-axis, with perfect alignment on every condensed ring. The crystalline cell is triclinic and each cell contains one coronene and one PTCDI-C6 molecule. It should be noted that there is a plane angle between the molecule stacking direction and the (100) face. Therefore, based on the XRD spectrum shown in Fig. 2b, one can calculate the plane spacing of (100) to be 3.56 Å. On the other hand, the distance between the D–A stacking of coronene and PTCDI can be calculated to be 3.30 Å (Fig. 2c) using the interatomic arrangement obtained from the crystalline structure. This calculated distance is consistent with the typical π–π stacking distance reported for other molecular assemblies.12,24,25
 | ||
| Fig. 2 (a) Triclinic cell showing the alternate stacking of coronene and PTCDI in the cocrystal. (b) XRD spectrum recorded on the cocrystal. (c) The π–π stacking between coronene and PTCDI-C6 with a spacing of 3.30 Å. (d) The hydrogen bonds between the perylene hydrogen and the imide oxygen of nearby PTCDIs. | ||
In the cocrystal, the molecular stacking configuration is quite different from what observed for the single component crystals of coronene and PTCDI-C6. According to our previous study,26 assembly of PTCDI-C6 molecules takes two orientations within one monoclinic crystal cell, as usually observed for other PTCDIs that have tertiary or quaternary carbon atoms directly linked to the imide nitrogen atoms.5,27 Within the PTCDI-C6 crystal, the stacking molecules twist by an appropriate angle to minimize the steric hindrance of cyclohexyl groups. But with the inserting of coronene molecules, the distance between the two PTCDI-C6 molecules was almost doubled, thus diminishing the steric hindrance of side groups. Therefore, the untwisted (parallel aligned) stacking between the PTCDI planes is enabled in the D–A cocrystal with the assistance from coronene. To this regard, the coronene molecule acts as both a spacer and interlocker that stabilizes D–A cocrystalline structure.
To further explore the intermolecular interaction in self-assembly, we performed solution phase photoluminescence and proton nuclear magnetic resonance (1H NMR) characterizations over the D–A assembly of coronene and PTCDI-C6 (Fig. S4†).28 Concentrated solutions of coronene and PTCDI-C6 (1 mM for each) were prepared in chloroform, with the former being colorless and the latter in orange color. When these two solutions were 1:1 mixed, the mixture turned to be dark red, indicating that the strong CT interaction between coronene and PTCDI-C6 exists in the solution state, which is a driving force to facilitate the molecular assembly.29 Meanwhile, the fluorescence of PTCDI-C6 was quenched upon mixing with coronene, as shown in Fig. S4a.† Comparative 1H NMR measurements over the deuterated chloroform (CDCl3) solutions of coronene, PTCDI-C6 and the 1:1 mixture demonstrate that the D–A assembly causes upfield shifting for the signals assigned to the hydrogens on the aromatic rings in both coronene and PTCDI-C6 (Fig. S4b†). Such shifting is likely due to the shielding from the aromatic ring current of the neighbouring coronene and PTCDI core stacking,30 which is consistent with the π–π stacking arrangement in the single crystal structure. So we attribute the strong D–A π–π stacking (in association with the CT interaction) is the major driving force for the cocrystal growth. Interestingly, upon D–A assembly, the original pair of roofed doublets of the central eight hydrogens of PTCDI-C6 (∼8.6 ppm) are further splitted into two separated doublet peaks. This indicates that half of the eight hydrogens are now in different chemical environment from the other half with the process of crystallization of coronene and PTCDI-C6, which is probably caused by the moderate interaction between these hydrogen atoms and the imide groups in PTCDI cores. To find more clear evidence, we turned back to the study on the cocrystal solid.
Generally, in addition to the π–π stacking, the hydrogen bonding also plays important role in formation of cocrystals.8,31–33 Indeed, moderate hydrogen bonds may exist in the coronene and PTCDI-C6 cocrystal. Due to the weak acidity of hydrogen on the perylene rings of PTCDI, they can form hydrogen bonds with the surrounding oxygen in the imide groups,34 for which the shortest distance between the hydrogen and oxygen atoms is 2.92 Å within the cocrystal, fitting into the typical “moderate” hydrogen bond length. To probe the existence of hydrogen bonding, the Fourier transform infrared (FTIR) spectroscopy measurement was performed on the coronene, PTCDI-C6 and the D–A cocrystal (Fig. S5†). PTCDI-C6 crystal can be considered as a reference because the twisted stacking of PTCDI-C6 molecules does not allow for effective hydrogen bonding between the hydrogen and imide oxygen, which are separated too far. As clearly shown in Fig. S5,† the stretching vibration band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O are red-shifted from νs(CO) = 1698.1 cm−1 and νa(CO) = 1659.4 cm−1 (measured for the D–A cocrystal) to νs(CO) = 1689.3 cm−1 and νa(CO) = 1655.4 cm−1 (measured for pure PTCDI-C6 crystal). Such shifting could be interpreted as the weakening of CO bond caused by the formation of hydrogen bond. On the other hand, the vibration of PTCDI ring (ν(ring) = 1593.9 cm−1) is very similar in both cocrystal and PTCDI-C6, that will act as an internal standard to exclude the possible environment difference between the two samples. Moreover, the weak intermolecular hydrogen bonding was also consistent with the previous 1H NMR measurement (Fig. S4b†), wherein the eight hydrogen atoms of the PTCDI core changed from the originally chemical environment (as evidenced by the pair of roofed doublets) to more different chemical environments (as evidenced by the separated doublet peaks).
O are red-shifted from νs(CO) = 1698.1 cm−1 and νa(CO) = 1659.4 cm−1 (measured for the D–A cocrystal) to νs(CO) = 1689.3 cm−1 and νa(CO) = 1655.4 cm−1 (measured for pure PTCDI-C6 crystal). Such shifting could be interpreted as the weakening of CO bond caused by the formation of hydrogen bond. On the other hand, the vibration of PTCDI ring (ν(ring) = 1593.9 cm−1) is very similar in both cocrystal and PTCDI-C6, that will act as an internal standard to exclude the possible environment difference between the two samples. Moreover, the weak intermolecular hydrogen bonding was also consistent with the previous 1H NMR measurement (Fig. S4b†), wherein the eight hydrogen atoms of the PTCDI core changed from the originally chemical environment (as evidenced by the pair of roofed doublets) to more different chemical environments (as evidenced by the separated doublet peaks).
As discussed above, the formation of the D–A cocrystal was mainly driven by the π–π stacking of coronene and PTCDI-C6. The π–π stacking with limited lateral offset usually leads to 1D crystal growth.35 Indeed, microsized fibril D–A cocrystal structures were fabricated via the solvent vapor diffusion induced self-assembly12 (see Experimental details). The microfibers obtained are roughly in the shape of belt (rectangular cross-section), with width of 30–60 μm, thickness of 10–30 μm, and length of over several hundred micrometers (Fig. 3, S6 and S7†). These 1D cocrystals provided a good platform to investigate the polarized photoluminescence at single fiber level, with the aim to determine the relative angle between the long axis of fiber and the cofacial stacking direction.26,36 Since the polarization is along the π–π staking direction, measuring the relationship between the luminescence intensity and its polarization will reveal the tilted angle of the molecular stacking (relative to the fiber long axis) as shown in Fig. 3a. The maximum luminescence was measured when the polarizer was aligned at 67° to the along axis of fiber, while the minimum luminescence was observed by rotating the polarizer 90° from the maximum position. The anisotropic ratio of the polarization (Imax/Imin) is calculated as ca. 3.
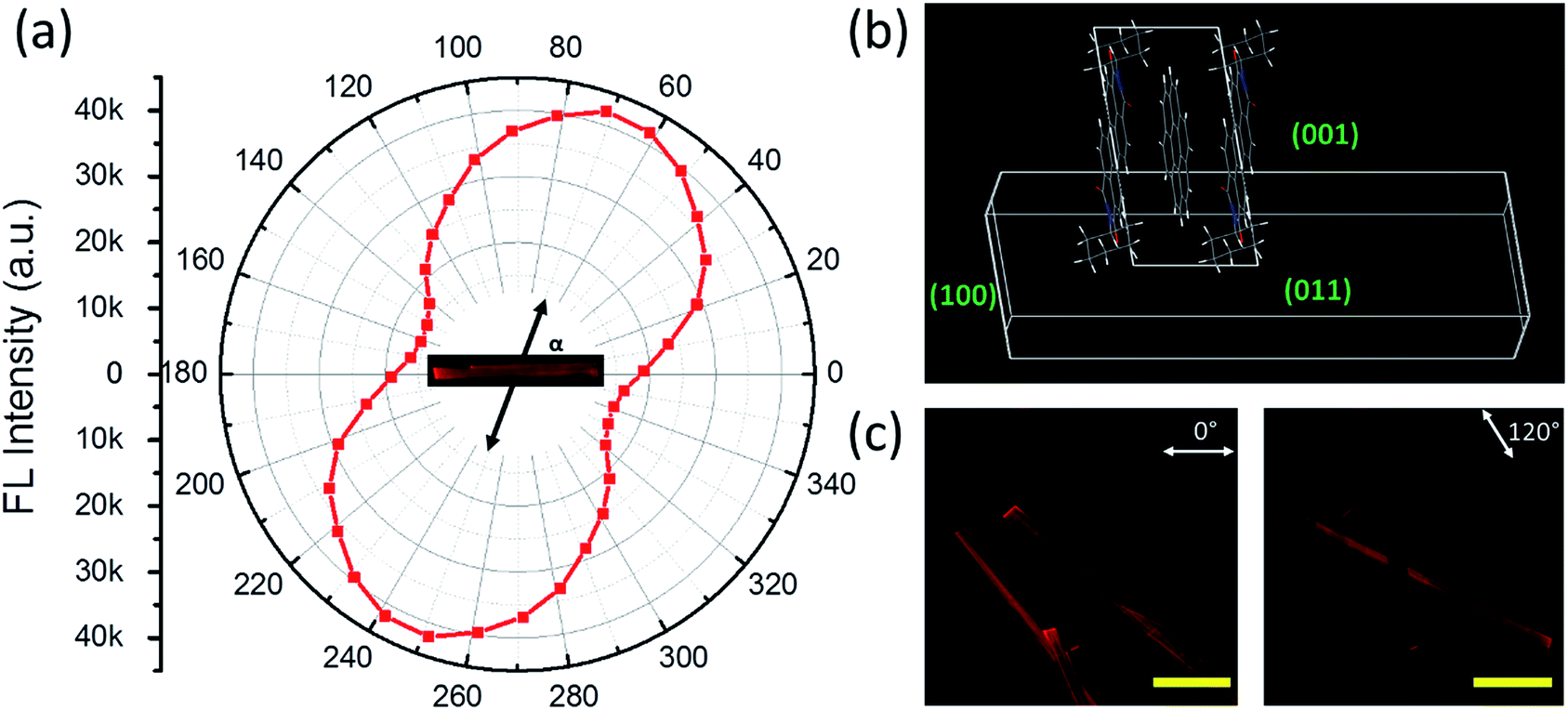 | ||
| Fig. 3 (a) Polar plot of fluorescence intensity measured on a cocrystal microfiber as a function of the angle (α) between the fluorescence polarization direction and the long axis of fiber. (b) Unit cell stacking pattern and exposing facets in a cocrystal microfiber. (c) Anisotropic fluorescence recorded over multiple microfibers under two different angles (marked) of polarized fluorescence light. Scale bar is 100 μm. | ||
A crystal growth simulation based on energy minimization was also performed based on the D–A cocrystal structure, from which the crystalline cell repeating pattern was obtained, as illustrated in Fig. 3b. From the top view, the direction of PTCDI-C6 stacking is consistent with the polarization measurement. The elongating growth direction (the left and right facets) can be confirmed as {100}, while the thickness (the top and bottom facets) and width direction (the front and behind facets) can be determined as {011}, {001}, respectively. As shown in the XRD data in Fig. 2b, these three groups of facets have relative high plane-to-plane spacing, so they would easily become the final outside facets. The facet ratios calculated from the facet energy (Table S2†) are also well consistent with the three-dimensional morphology of the microfibers (width vs. thickness vs. length), implying that the growth of the cocrystal was mainly a thermodynamically controlled process.37 Additionally, based on this simulation, the angle of the cofacial stacking direction with respect to the long axis of fiber is consistent with the measured direction with maximum photoluminescence, which was 67°. Polarized luminescence was also measured simultaneously over multiple fibers as shown in Fig. 3c and S6.† With the rotation of polarizer different fibers showed different intensity of luminescence depending on the polarization direction, and after calculation for each microfiber, their directions with the maximum photoluminescence were between near to 67°. Furthermore, the well-defined 1D shape and the strong rigidity of the D–A cocrystal allow for fabrication of large-area array of aligned microfibers, which are usually desired for application in electronic devices.38 For example, by evaporating the solvent at a controlled rate from the 1:1 D–A solution in chloroform, we could obtain highly aligned microfibers, and the array also had highly polarized fluorescence with an anisotropic ratio over 2.5 (Fig. S7†).
Conclusions
In summary, we have successfully designed and fabricated a D–A cocrystal through molecular self-assembly of coronene and a PTCDI molecule in a 1:1 chloroform solution. Driven by the π–π stacking interaction, the D–A cocrystal grows mostly along one dimension, forming 1D microstructures. The crystalline structure is consisted of triclinic cell, within which the PTCDI and coronene molecules stack alternately at a titled angle of 67° with respect of the long axis of the microfiber. Such 1D dominant crystal demonstrated significant polarization of photoluminescence, with anisotropic ratio about 3. Due to the strong CT interaction between coronene and PTCDI, the cocrystal also showed clear red-shift absorption and photoluminescence bands. Additionally, a simple solution method could be used to directly fabricate the D–A cocrystal into large area alignment of the microfibers, which are conducive to further fabrication into electronic devices. Combination of these features will open great potential for applying such D–A cocrystal materials, along with further study of the basic optoelectronic properties and relationship with the intermolecular arrangement.
Experimental
Materials
PTCDI-C6 was synthesized following our previous published method.26 Coronene (97%) and the synthesis precursor of PTCDI-C6 (perylene-3,4,9,10-tetracarboxylic dianhydride and cyclohexylamine), and solvents (diethyl ether, diisopropyl ether, and chloroform) were purchased form Sigma-Aldrich, and used without further treatment.Cocrystal growth
Chloroform solution containing coronene (1 mmol L−1) and PTCDI-C6 (1 mmol L−1) was prepared as the stock solution. For the large cocrystal growth in the single crystal XRD measurement, 5 mL of this solution was put into a 10 mL glass vial, which was then put in a large container where saturated diisopropyl ether vapor was maintained. Such slow vapor diffusion into the chloroform solution leads to growth of the cocrystal in a size that is sufficiently large for single crystal XRD measurement. The similar method was also used for making the microsized fibers (like those shown in Fig. 3) but using diethyl ether as the solvent vapor source instead. To prepare the cocrystal microfibers array (Fig. S7†), 8 mL of the stock solution was added to a 10 mL glass vial. A glass or silicon substrate was put inside the vial (with the substrate parallel to the vial wall). As the solvent evaporated slowly, the microfibers formed with the growth along the direction of solvent front moving.38Spectral measurement
Chloroform solutions of coronene (1 mmol L−1) and PTCDI-C6 (1 mmol L−1), and solid samples of coronene, PTCDI-C6 and the D–A cocrystal on the quartz slides were prepared for UV-vis absorption and fluorescence spectral measurement. The absorption was measured with an Agilent Cary 100 series UV-vis spectrophotometer, and the fluorescence (including the excitation spectra) was measured with an Agilent Cary Eclipse Fluorescence Spectrophotometer. The temperature-dependent fluorescence was measured with Ocean Optics USB4000 detector and the sample was excited by a 532 nm 10 ns pulse laser. Fluorescence quantum yield was measured with the cocrystal powders deposited on a polytetrafluoroethylene film using a Hamamatsu Absolute PL Quantum Yield Spectrometer (C11247) via the integrating sphere method. The 1H NMR spectra were recorded on a Varian Unity 300 Spectrometer, for which CDCl3 solutions of coronene and PTCDI-C6 (both ca. 2 mmol L−1) and their 1:1 mixture were used. FTIR spectra of coronene, PTCDI-C6 and the D–A cocrystal were obtained using a Nicolet Magna 750 Fourier transform infrared spectrometer equipped with a liquid-nitrogen-cooled mercury cadmium telluride detector.
Polarized fluorescence measurement
The fluorescence polarization was measured with a Leica DMI4000B inverted microscope, which was equipped a rotatable linear polarizer to polarize the fluorescence beam after the excitation by a non-polarized green light from a mercury lamp. The spectra of the polarized fluorescence was measured by a Princeton Instrument SpectraPro 2300i monochromator and PIXIS 400 CCD for spectra measurement.Acknowledgements
We thank the help on FTIR measurement from Dr Marc Porter, and the help on the structure solution and refinement of the D–A cocrystal from Dr Atta M Aarif. This work was supported by the funding from the NSF (CHE 0931466 and CBET 1502433), DHS Science and Technology Directorate (2009-ST-108-LR0005) and the USTAR Program.Notes and references
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- B. Carsten, J. M. Szarko, H. J. Son, W. Wang, L. Lu, F. He, B. S. Rolczynski, S. J. Lou, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2011, 133, 20468–20475 CrossRef CAS PubMed.
- J. Ferraris, D. Cowan, V. T. Walatka and J. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- S. K. Park, S. Varghese, J. H. Kim, S. J. Yoon, O. K. Kwon, B. K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- Y. Su, Y. Li, J. Liu, R. Xing and Y. Han, Nanoscale, 2015, 7, 1944–1955 RSC.
- A. S. Tayi, A. Kaeser, M. Matsumoto, T. Aida and S. I. Stupp, Nat. Chem., 2015, 7, 281–294 CrossRef CAS PubMed.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- N. Blagden, D. J. Berry, A. Parkin, H. Javed, A. Ibrahim, P. T. Gavan, L. L. De Matos and C. C. Seaton, New J. Chem., 2008, 32, 1659–1672 RSC.
- C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J.-L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS.
- K. Y. Law, Chem. Rev., 1993, 93, 449–486 CrossRef CAS.
- B. A. Gregg, J. Phys. Chem. B, 2003, 107, 4688–4698 CrossRef CAS.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS PubMed.
- A. L. Briseno, S. C. B. Mannsfeld, C. Reese, J. M. Hancock, Y. Xiong, S. A. Jenekhe, Z. Bao and Y. Xia, Nano Lett., 2007, 7, 2847–2853 CrossRef CAS PubMed.
- C. Wang, B. R. Bunes, M. Xu, N. Wu, X. Yang, D. E. Gross and L. Zang, ACS Sens., 2016, 1, 552–559 CrossRef CAS.
- S. Prasanthkumar, S. Ghosh, V. C. Nair, A. Saeki, S. Seki and A. Ajayaghosh, Angew. Chem., Int. Ed., 2015, 54, 946–950 CrossRef CAS PubMed.
- J. López-Andarias, M. J. Rodriguez, C. Atienza, J. L. López, T. Mikie, S. Casado, S. Seki, J. L. Carrascosa and N. Martín, J. Am. Chem. Soc., 2015, 137, 893–897 CrossRef PubMed.
- L. Bu, T. J. Dawson and R. C. Hayward, ACS Nano, 2015, 9, 1878–1885 CrossRef CAS PubMed.
- J. He, D. M. Agra-Kooijman, G. Singh, C. Wang, C. Dugger, J. Zeng, L. Zang, S. Kumar and C. S. Hartley, J. Mater. Chem. C, 2013, 1, 5833–5836 RSC.
- H. Huang, C.-E. Chou, Y. Che, L. Li, C. Wang, X. Yang, Z. Peng and L. Zang, J. Am. Chem. Soc., 2013, 135, 16490–16496 CrossRef CAS PubMed.
- N. Wu, C. Wang, B. R. Bunes, Y. Zhang, P. M. Slattum, X. Yang and L. Zang, ACS Appl. Mater. Interfaces, 2016, 8, 12360–12368 CAS.
- J. Xiao, H. Yang, Z. Yin, J. Guo, F. Boey, H. Zhang and Q. Zhang, J. Mater. Chem., 2011, 21, 1423–1427 RSC.
- G. Balaji, T. S. Kale, A. Keerthi, A. M. D. Pelle, S. Thayumanavan and S. Valiyaveettil, Org. Lett., 2011, 13, 18–21 CrossRef CAS PubMed.
- X. Zhan, Z. Tan, E. Zhou, Y. Li, R. Misra, A. Grant, B. Domercq, X. Zhang, Z. An, X. Zhang, S. Barlow, B. Sippelen and S. R. Marder, J. Mater. Chem., 2009, 19, 5794–5803 RSC.
- G. Giri, E. Verploegen, S. C. B. Mannsfeld, S. Atahan-Evrenk, D. H. Kim, S. Y. Lee, H. A. Becerril, A. Aspuru-Guzik, M. F. Toney and Z. Bao, Nature, 2011, 480, 504–508 CrossRef CAS PubMed.
- S. C. B. Mannsfeld, M. L. Tang and Z. Bao, Adv. Mater., 2011, 23, 127–131 CrossRef CAS PubMed.
- Y. Che, X. Yang, K. Balakrishnan, J. Zuo and L. Zang, Chem. Mater., 2009, 21, 2930–2934 CrossRef CAS.
- K. J. Hartlieb, L. S. Witus, D. P. Ferris, A. N. Basuray, M. M. Algaradah, A. A. Sarjeant, C. L. Stern, M. S. Nassar, Y. Y. Botros and J. F. Stoddart, ACS Nano, 2015, 9, 1461–1470 CrossRef CAS PubMed.
- C. Zhan, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 4625–4628 CrossRef CAS PubMed.
- T. Aoki, H. Sakai, K. Ohkubo, T. Sakanoue, T. Takenobu, S. Fukuzumi and T. Hasobe, Chem. Sci., 2015, 6, 1498–1509 RSC.
- J.-M. Han, N. Wu, B. Wang, C. Wang, M. Xu, X. Yang, H. Yang and L. Zang, J. Mater. Chem. C, 2015, 3, 4345–4351 RSC.
- D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 1106–1123 CAS.
- Q. Liao, H. Fu, C. Wang and J. Yao, Angew. Chem., Int. Ed., 2011, 50, 4942–4946 CrossRef CAS PubMed.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932–1943 Search PubMed.
- Z. Liu, G. Liu, Y. Wu, D. Cao, J. Sun, S. T. Schneebeli, M. S. Nassar, C. A. Mirkin and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 16651–16660 CrossRef CAS PubMed.
- S. Chen, P. Slattum, C. Wang and L. Zang, Chem. Rev., 2015, 115, 11967–11998 CrossRef CAS PubMed.
- A. Datar, K. Balakrishnan, X. Yang, X. Zuo, J. Huang, R. Oitker, M. Yen, J. Zhao, D. M. Tiede and L. Zang, J. Phys. Chem. B, 2006, 110, 12327–12332 CrossRef CAS PubMed.
- L. Huang, Q. Liao, Q. Shi, H. Fu, J. Ma and J. Yao, J. Mater. Chem., 2010, 20, 159–166 RSC.
- F. L. Geyer, A. Pun, D. Hanifi, U. H. F. Bunz and Y. Liu, J. Mater. Chem. C, 2013, 1, 6661–6666 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1501468. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra25447k |
| This journal is © The Royal Society of Chemistry 2017 |