
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering-scale superlubricity of the fingerprint-like carbon films based on high power pulsed plasma enhanced chemical vapor deposition
Zhenbin
Gong
ab,
Jing
Shi
ab,
Wei
Ma
a,
Bin
Zhang
a and
Junyan
Zhang
*a
aState Key Laboratory of Solid Lubrication, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China. E-mail: zhangjunyan@licp.cas.cn
bUniversity of Chinese Academy of Sciences, Beijing, 10049, China
First published on 14th December 2016
Abstract
It has been a great challenge to achieve superlubricity on an engineering scale. In this study, macro superlubricity was realized by fingerprint-like carbon (FP-C:H) films that were prepared by a high power pulsed plasma enhanced chemical vapor deposition technique. The macro superlubricity occurred under a wide range of test conditions, with a super low friction coefficient of 0.0016 in dry air. The unique structure and properties of the graphene layers made it capable not only to lower the shearing stress but also efficiently achieve superlubricity, following reorganization mechanics. High-resolution transmission electron microscopy (HRTEM) and Raman spectroscopy revealed the nanostructure evolution of the wear debris. Surprisingly, a kind of multistorey graphene nanoparticles were generated in the wear debris and the humidity played an important role in the formation of these particles. Moreover, the nanostructures of these particles directly affected the friction coefficients at different humidity values. It can be demonstrated that the graphene nanoparticles were the major reason for the super lubrication of fingerprint-like carbon films, achieving incommensurate and rolling contacts. An engineering applicable method combined with the unique superlubricity properties of fingerprint-like carbon could offer an exciting opportunity to realize long-sought applications in vehicles, turbines, and manufacturing equipment.
1. Introduction
Both one-third of the world's primary energy consumption and four-fifth of the mechanical components failures are contributed to friction.1,2 Therefore, friction reduction is particularly important for the environment and sustainable development.3 High requirements for superlubricity materials and technology have been put forward along with the development of vehicles, turbines, and manufacturing equipment. Although it is believed that superlubricity can save energy and extend the life of the moving parts, both technique and materials towards this have not yet been well-developed. In the past few decades, two kinds of materials showed an ultra-low friction coefficient and became interesting research topics in the field of superlubricity: one is two-dimensional materials,4–6 such as graphene, graphite, and graphite/C60,7–9 where the incommensurate contact can significantly reduce the friction and wear under specific conditions, and this unique friction state regarding the atomic lattice structures is called the “structural lubricity”; other one is the carbon based thin films, such as diamond like carbon and ultra nanocrystalline diamond films (friction coefficient ∼ 0.001).10,11 However, nearly all the experimental superlubricity has been obtained on microscope or under specific conditions (vacuum or dry conditions), which are obstacles towards its engineering applications since the superlubricity systems diminish most of its properties on the bulk scale or under atmosphere.12–14Graphene exhibits the potential to achieve ultra-low friction, expected as an ideal candidate for the lubrication.15 Recently, macroscopic superlubricity was realized by graphene and nanodiamonds at the macroscale. The graphene sheets exhibited self-organization behavior at the sliding interface, forming nanoscrolls to create macroscopic scale superlubricity.16 A nanoscroll reduces the adhesion between the contact surfaces and achieves incommensurate contact, greatly reducing the friction and leading to structural superlubricity. However, in practical applications, thin films need to have a comprehensive performance, with good mechanics, stability, and high binding force. As a result, it remains an open question to design a solid surface at the nanoscale to sustain superlubricity at the engineering scales.
To bridge the gap between superlubricity and practical application, great efforts have been made to coat thin films on the component surfaces with various materials and structures.17 Vacuum coating is one of the most advanced technologies in the scientific research and industrial engineering, which makes it easy to achieve the comprehensive performance.1 Most importantly, adjusting the preparation process can control the nanostructure of thin films, which is the prerequisite for structural superlubricity. Generally, solid superlubricity mainly relies on the formation of incommensurate contact, which can be achieved by a variety of carbon nanostructures, such as graphite,7–9 graphene,12 nanodiamonds,16 and nanotubes.18,19 To date, it has been reported that thin films with novel nanostructure could be prepared with macro superlubricity properties through plasma vacuum deposition technology.20–22 However, extreme conditions or high energy preparation methods are necessary to synthesize these nanostructures; for example, graphene,23,24 nanotube,25,26 nanodiamonds,27 and fullerene28 can only be fabricated at high temperature, with high energy ions states, or with catalysts, and nanocrystallites-embedded amorphous carbon require methods such as modified arc techniques.29–33 Thus, the growth energy greatly impacts the formation of the nanostructured carbon. Moreover, for graphene, high growth energy can be reached via heating or high energy excitation and, therefore, involves high temperature (from 500 °C to more than 1000 °C) or methods such as electron irradiation in the electron cyclotron resonance, mass selected ion beam deposition, arc-discharge, and microwave plasma enhanced chemical vapor deposition.23,34–41 This demonstrates that the high energy state can promote the formation of graphene-like sheets. High-power pulsed power supply, exciting ions (near arc discharge) with very high power densities and ion energy,42,43 could be used to prepare the graphene-like nanostructures.
In this study, a type of fingerprint-like carbon films (FP-C:H) with curved graphene layers were deposited by a high power pulsed-plasma enhanced chemical vapor deposition technique (HiP-PECVD). It was proven that the films possessed stable superlubricity under a wide range of test conditions: counterpart balls from Al2O3, Si4N3 to steel, load from 2 N to 20 N in dry air or with low humidity. It is believed that the superlubricity originates from the self-organization of graphene sheets to form graphene nanoparticles (GNPs), which offers an incommensurate and rolling contact. In addition, the simple method combined with the unique superlubricity properties offers an exciting opportunity to realize the long-sought applications in vehicles, turbines, and manufacturing equipment.
2. Experimental
Film preparation
Nanostructure carbon films were synthesized using HiP-PECVD. The base pressure of the deposition chamber was about 1 × 10−4 Pa. The samples were deposited on Si (100) in a flow of CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:1 in a mixture of methane (99.99%) and hydrogen (99.99%) discharge, under work pressure at 30 Pa. The power supply (JX-HPMS, JINXIN, CHINA) operated in a constant voltage mode under a negative voltage of −2000 V with a peak current of 80 A (pulse frequency: 350 Hz). The duty cycle was 0.05, and accordingly, the maximum power was 160 kW, whereas the average power was 8 kW. The samples were mounted at a distance of 5 cm from the cathode.
:H2 = 1:1 in a mixture of methane (99.99%) and hydrogen (99.99%) discharge, under work pressure at 30 Pa. The power supply (JX-HPMS, JINXIN, CHINA) operated in a constant voltage mode under a negative voltage of −2000 V with a peak current of 80 A (pulse frequency: 350 Hz). The duty cycle was 0.05, and accordingly, the maximum power was 160 kW, whereas the average power was 8 kW. The samples were mounted at a distance of 5 cm from the cathode.
HRTEM samples preparation
The high-resolution transmission electron microscope (HRTEM) samples were grown on freshly cleaved NaCl wafers with a thickness about 20 nm. To avoid the metal contamination, before the TEM sample deposition, the substrate holder and vacuum chamber wall were coated by carbon films. The wear debris samples were prepared using a micro grid to polish the wear scar of the counterpart balls after friction testing.Thickness measurement
The thickness of the film was obtained by a scanning electron microscope (JEOL JSM-6701F) from their cross-section samples.Structure characterizations
Nanostructure of the thin films and wear debris were analyzed using a HRTEM Tecnai-G2 F30 (FEI, US) and Tecnai-G2 F20 (FEI, US), respectively. The graphene domain information was studied using atomic force microscopy (AFM Nanoscope Maltimode 8.0, Bruker Co. GER). In addition, further information regarding the structural arrangement was studied using a micro Raman spectrometer (LABRAM HR 800) at a wavelength of 532 nm (2.3 eV). Film composition and bonding information were analyzed by XPS (VG ESCALAB 210) using an Al-Kα (1486.5 eV) X-ray radiation.Nanoindentation tests
The mechanical properties of the FP-C:H films were measured by nanoindentation (Hysitron Ti-950) with a trigonal Berkovich diamond tip. The maximum indentation depth was controlled at about 50 nm (1/10 of the film thickness). The elastic recovery R is defined as follows:| R = (dmax − dres)/dmax × 100% | (1) |
Tribological characterization
The tribological performances of the specimens were evaluated by the ball-on-disk reciprocating friction testing machine under dry, 10%, 20%, 30%, 40%, and 50% relative humidity atmospheres. The counterpart balls were Al2O3, ZrO2, Si4N3, and steel with a diameter of 5 mm. The loading forces of the friction tests were 20 N, 15 N, 10 N, 5 N, and 2 N, respectively. The sliding diameter was 8 mm, and the frequency was 10 Hz.3. Results and discussion
Fingerprint-like carbon (FP-C:H) with a thickness of about 700 nm were deposited on the silicon substrates. The nanostructure of the carbon films is shown in Fig. 1. The film has a very attractive and unique nanostructure (Fig. 1a), which can be described as distorted multistorey graphene arrangements in an amorphous body. The distance between two graphene sheets was about 0.34 nm. Note that these patterns are very similar to those of a human fingerprint (inset picture), so it was named as fingerprint-like carbon (FP-C:H). Further, AFM phase imaging under tapping mode was used to study the graphene arrangement features and distributions. Since the phase image of AFM can distinguish the surface stiffness variations associated with Young's modulus changes, this can be used to identify the nanocluster domains in the film.44Fig. 1b shows an AFM phase image of a fingerprint-like carbon film deposited on a silicon wafer. It is obvious that the film had a special nanocomposite structure with graphene domains (soft) dispersed in an amorphous network (hard). | ||
| Fig. 1 (a) High-resolution transmission electron images of the FP-C:H films. Insets: carbon fingerprint and human fingerprint pattern. (b) An AFM phase image of the fingerprint-like carbon film deposited on a silicon wafer. | ||
Raman spectroscopy, which can identify various carbon nanostructures, bonding structure, vibration mode and order degree, is a powerful tool to analyze the carbon-based materials.45 The FP-C:H films (Fig. 2a) show a typical feature spectrum of amorphous carbon from 1000 cm−1 to 1800 cm−1 at 633, 532, and 325 nm Raman exciting laser wavelengths. The main peak was decomposed into D and G bands by guess mode, where 0.62, 0.79, and 0.99 are the ID/IG values for 633, 532, and 325 nm excitation wavelengths, respectively. The low values of ID/IG indicate that the FP-C:H films had a high sp2 content. The size of the sp2 cluster (La) was calculated by the Tuinstra–Koenig relation as follows:46
 | (2) |
 | (3) |
 | ||
| Fig. 2 (a) Raman spectra of the FP-C:H films with 633, 532, and 325 nm Raman excitation wavelengths. (b) The corresponding fitted Raman spectra of the FP-C:H films at 532 nm from 2400 to 3500 cm−1. (c) The deconvolution of the high-resolution XPS spectra of the C1s peak for FP-C:H films. | ||
Therefore, the equation can be simplified as follows:47
 | (4) |
 | ||
| Fig. 3 (a), (b) and (c) The friction coefficient curves as a function of rubbing cycles at different humidity against Al2O3, Si4N3, and steel ball, respectively. (d) Friction coefficient curves of the FP-C:H films in a dry air atmosphere under a load from 2 N to 20 N, the inset shows the friction coefficient as a function of load. | ||
To further understand the nanostructure of FP-C:H, the elements composition and chemical structures were studied by XPS. Fig. 2c indicates the deconvolution of the XPS spectrum of the FP-C:H film. The C1s spectrum consists of four features of binding energy values obtained at 284.5, 285.2, 286.5, and 288.5 eV, corresponding to sp2, sp3, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and the full width at half maximum (FWHM) were 1.0, 1.0, 1.4, and 2.3 eV, respectively.56–58 The oxygen peaks were originated from the surface contamination when the FP-C:H film was exposed to air.59 Combining the results of HRTEM, Raman, and XPS, it can be concluded that graphene layers with a fingerprint-like pattern have been formed within the as-deposited carbon films.
O, and the full width at half maximum (FWHM) were 1.0, 1.0, 1.4, and 2.3 eV, respectively.56–58 The oxygen peaks were originated from the surface contamination when the FP-C:H film was exposed to air.59 Combining the results of HRTEM, Raman, and XPS, it can be concluded that graphene layers with a fingerprint-like pattern have been formed within the as-deposited carbon films.
The mechanical properties of the carbon thin film had a very significant effect on its friction performance and engineering applications.60 As listed in Table 1, the FP-C:H films exhibited a super-high elastic recovery (R) of 84.6%, which indicates a minimal plastic deformation during the indentation process. It also exhibited a high hardness of up to 22.8 GPa. Arguably, the three dimensions buckled and curved graphite layers gave rise to the high hardness and elastic recovery of the FP-C:H carbon films.
| Indentation depth, nm | Hardness, GPa | Elastic module, GPa | Elastic recovery, % | |
|---|---|---|---|---|
| Test | 45–52 | 20.2–24.6 | 140.1–162.2 | 82–87 |
| Average | 49 | 22.8 | 154.2 | 84.6 |
Moreover, the attractive macroscopic superlubricity properties have been observed for the FP-C:H carbon films. The friction behaviors were tested under dry, 10%, 20%, 30%, 40%, and 50% humidity atmospheres with different dual balls and loads (Fig. 3). The ambient temperature was about 20 °C, and the sliding speed was about 160 mm s−1. An extremely low friction coefficient below 0.0016 was observed for the FP-C:H films sliding against the Al2O3 balls in a dry environment. Our experimental studies also confirmed that the stable superlubricity regime occurred under a wide range of test conditions: counterpart balls changed from Al2O3, Si4N3 to steel, load from 2 N to 20 N, and atmosphere from dry air to low-humidity air. For example, the Al2O3/FP-C:H friction couple had a super low friction coefficient under low-humidity air (friction coefficient was less than 0.01 when the humidity was below 30%). At the same time, macroscale superlubricity could be achieved under a load ranging from 2 N to 20 N. From the friction curves (Fig. 3a–c), it was found that for all the friction pairs, an increase in the humidity led to an increase in the friction. The friction mechanism is very important for understanding the superlubricity properties of the thin films. Previous studies have shown that superlubricity can be achieved by the hydrogenated and sulfur-doped hydrogenated carbon films, where the ultra-low friction originates from the passivation species in the test environment, such as H or S–H bonding.61,62 However, for FP-C:H films, there is no highly passive H bonds due to the oxygen enrichment41 and though O–H has a higher bond energy than that of S–H63 because the oxidation of carbon is preferred. Therefore, it seems reasonable to surmise that the special nanostructure in the FP-C:H films greatly enhanced the tribological properties.
To explore the ultra-low friction mechanism of the FP-C:H films, the wear debris and wear scar were monitored by Raman spectroscopy. The Raman spectra were obtained at different positions at one time for each sample, as shown in Fig. 4a. The results (Fig. 4b) indicated that the structural transformation of the freshly exposed surface was not obvious; however, the wear debris showed significant structural changes reflected by the Raman spectra red shift, and the area ratio of I2D/ID+G + I2D increased with a decrease in the humidity. According to the abovementioned discussion, the area of 2D range can be regarded as the contents of graphene structure. Fig. 4c shows the friction coefficients and 2D range proportion as a function of humidity. The results of Raman spectroscopy demonstrated that the wear scar had a similar structure as the intrinsic films, whereas the interface structure evolution and bonding recombination mainly occurred in the wear debris. For the same friction experiment, the more dry the environment, the more its conducive for the formation and accumulation of graphene structure.
 | ||
| Fig. 4 (a) Microscopic images of the wear debris and wear scar. (b) Raman spectra of the original films, wear scar, and the wear debris (under dry, 10%, 20%, 30%, 40%, and 50% related humidity, respectively). (c) Friction coefficient and 2D peak as a function of humidity. | ||
The FP-C:H films suffered from significant wear during sliding, however, the structural changes occurred mainly in the wear debris (Fig. 4a and b). It was found that the superlubricity could be achieved under a wide range of test conditions, but was influenced by the related humidity (HR). Therefore, an understanding of the humidity-controlled wear debris evolution could further explore the underlying mechanism of the superlubricity. To study the nanostructure of the wear debris on the Al2O3 balls under different humidity values, a more detailed analysis of the wear debris formed during the superlubricity regime in dry air was carried out by HRTEM. Fig. 5 shows the nanostructure transformation of the wear debris. Surprisingly, multilayer graphene nanoparticles (GNPs) were observed in the wear debris. This unique phenomenon is likely to be a key to understanding the origin of an ultra-low friction coefficient of the FP-C:H films. In dry environments, the GNPs indicated a perfect spherical shell structure with fewer defects (Fig. 5a). Under the 10% relative humidity (Fig. 5b), the multilayer shell structure could still be maintained, but with more defects. When the humidity was increased to 20% (Fig. 5c), the laminated shell structure distortion and lamella spacing distribution became wider and was mixed with an amorphous structure (Fig. 5d). Under 30% humidity, the shell structure of the GNPs was broken, the defects increased, and an amorphous structure was introduced. At 40% humidity, the multilayer shell structure disappeared in the wear debris and only short curved graphene sheets could be observed (Fig. 5e). When the humidity was increased to 50%, the GNPs showed a typical amorphous structure with short-range fingerprint-like graphene sheets (Fig. 5f). The wear debris was composed of spherical nanoparticles, which originated from the multilayer graphene. The decrease in the particle radius (Fig. 5, inset table) was accompanied by the destruction of the shell structure, which increased the resistance of friction. The graphitic layers with a lamellar spacing of 0.32–0.40 nm were not perfectly conformal and did not have a continuous structure. Obviously, the nanostructure transformation of wear debris along with humidity was in well accordance with the friction coefficients. By analyzing the Raman spectra and HRTEM images, it was determined that the superlubricity of the FP-C:H films originated from the evolution of GNPs.
 | ||
| Fig. 5 HRTEM images of the wear debris: (a), (b), (c), (d), (e) and (f) under 0%, 10%, 20%, 30%, 40%, and 50% humidity, respectively, at 10 N and 10 Hz. Inset table, the average diameter of the graphene nanoparticles at different humidity values. | ||
Based on the results of tribology, HRTEM, and Raman, it was concluded that under a certain contact pressure, humidity controls the formation of GNPs, which further influence the friction behavior. In other words, the superlubricity of the FP-C:H films is related to the formation of GNPs at the counterparts interfaces, thus, revealing that the formation mechanism can help us to better understand the superlubricity.
In the present study, we proposed that the formation mechanism of GNPs is the self-organization of graphene sheets, which can be divided into two steps, nucleation and growth. The nucleation of the GNPs core is similar to the reaction pathway for the transformation of graphene-to-fullerene since the molecular cage of fullerene is more stable than that of graphene.63–65 However, the driving force of the process is not high temperature but the friction shear force. A possible nucleation and growth mode is given in Fig. 6. The nucleation process can be divided into four stages: (1) at the initial stage, the dual surface and the film have strong coupling, resulting in a strong wear and a large number of wear debris. The graphene layers (Fig. 6a) peeled off from the bulk body and directly contacted with the dual surface (Fig. 6f); (2) subsequently, these graphene sheets (Fig. 6b) (1) are folded and coiled under the shear stress (Fig. 6g); (3) with the development of a friction process, these short-range graphene fragments are coiled to form a cage-like structure under the action of periodic shearing forces (Fig. 6h); and (4) after more friction cycles, the carbon cage-like structure becomes more regular (Fig. 6i), where the positions 2 and 3 (Fig. 6c) mark a monolayer and a double layer, respectively. The single wall carbon cage diameter was about 0.7 nm, which has a radius similar to that of C60 (0.71 nm).
 | ||
| Fig. 6 (a) HRTEM images of the original FP-C:H films. (b)–(d) Wear debris of 600, 1200, and 6000 friction cycles, at 10 N and 10 Hz. (e) Raman spectra of the original films and wear debris of 600, 1200, and 6000 friction cycles, respectively. (f)–(m) Schematic of the nucleation and growth model for GNPs. | ||
The growth mechanism can be inferred from the HRTEM images of the wear debris. The defected graphene layers (soft) are easy to package a fullerene core (hard), which is similar to the mechanism reported by A. V. Sumant et al.16 The growth model of the GNPs was similar to that of the snowball effect. When the friction process tends to reach a balance, the thermodynamic non-equilibrium state of the FP-C:H films tends to reach an equilibrium state. A large class of GNPs could be formed after 3000 friction cycles (Fig. 6d). Few GNPs layers (position 4) could be found on the edge of a big particle, which can be seen as an evidence for this form of nucleation and growth pattern. Raman spectra of the original films and wear debris of 600, 1200, and 6000 friction cycles also confirmed this growth model (Fig. 6e) since the I2D/ID+G + I2D values increased accordingly. Fig. 6j–m are a possible growth model. Due to the spontaneous reduction of the surface energy, the graphene sheets tended to wrap around the carbon cage core to reduce the surface area (Fig. 6j and k). Under the effect of shear stress, the bonding recombination occurred at the interface, which finally leads to the formation of multilayer GNPs (Fig. 6l and m).
Theoretical study of the graphene–fullerene transformation confirmed that the formation of defects at the edge of graphene is the crucial step.63 To understand why water molecules can control the self assembly process, its molecular structure on the graphene edges should be further explored. The water molecules are made up of oxygen and hydrogen, with respective electro negativities of 3.44 and 2.20 (Fig. 7a). The dipoles from each of the two bonds (black arrows) add together to make the overall molecule polar (gray arrow) with a dipole moment of magnitude p = 6.20 × 10−30 C m.66 Graphene is always found to be p-doped as a result of H2O adsorption from air.67 Water molecules exhibit higher activation energies than those of the non-polar adsorbates (O2, N2, and Ar) and tend to form hydrogen bonds, which can be attributed to stronger adsorbate–adsorbate interactions.68 Under dry environments, unsaturated dangling bonds of graphene had a high reaction activity, and the hybridization was easy to occur on the edge of graphene fragments (Fig. 7b). When water molecules are involved in the friction process, some of the dangling bonds are saturated by the water molecules, which blocks the hybridization of the graphene fragment (Fig. 7c). Under high humidity conditions (Fig. 7d), the water molecules almost saturate the dangling bonds of the lamellar surfaces, which limits the nucleation and growth of GNPs.
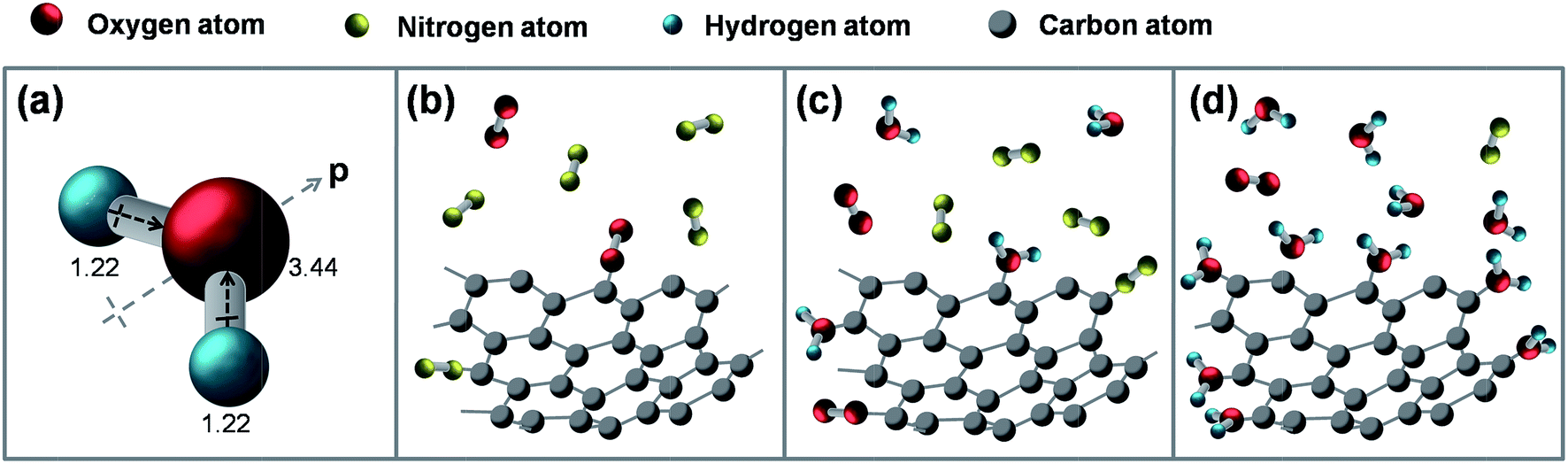 | ||
| Fig. 7 The structure of water molecules: (a) molecular adsorption models of the graphene fragments at different humidity values: (b) dry atmosphere, (c) low humidity, and (d) high humidity. | ||
It is expected that the multistorey spherical nanostructures would provide wonderful tribological properties compared to those of the amorphous form of the solid lubricants.16,18 Combined with the HRTEM results, it is speculated that the GNPs in the debris could constitute an incommensurate contact surface because the number of atoms in the GNPs are not the same as in the FP-C:H carbon surfaces. Furthermore, it is known that the coefficients of rolling friction are generally 102 to 103 times lower than those of the sliding friction for the corresponding materials.69 This is obvious because most of the nanoparticles obtained through the friction process have a regular spherical shape. In addition, these particles have a chemically inert surface, which reduces the couple of the dual interface. Actually, even under high humidity values up to 30%, wear debris can only form a multilayer structure, and the low shear-strength properties can be maintained during the tribological testing. This is due to the creep deformation of the curved layered structure in the wear debris. The frictionless property disappears only when the wear debris tends to be amorphous (humidity reaches 40% or more).
4. Conclusions
In summary, FP-C:H films were fabricated by the HiP-PECVD technique at room temperature. The films have fingerprint-like structures, which endow high hardness, high elastic recovery, and an ultra-low friction coefficient (μ = 0.0016) both in dry atmosphere and low humidity. The FP-C:H films could meet the requirements of the engineering scales for superlubricity, such as load, humidity, and dual materials, which overcome the shortcoming of the existing technology that superlubricity can only be acquired at microscale or under special conditions (vacuum or dry atmosphere). At the same time, the mature method (PECVD) reduces the cost and technical difficulty in the industry applications and enhances the stability and reliability of this superlubricity technology. For friction mechanism, it is believed that the superlubricity originates from the self-organized GNPs in the wear debris, with lower shear-strength properties, and offers incommensurate and rolling contact. The stress-induced GNPs formation by the interface is the possible reason for the existence of superlubricity in the dry and low humidity conditions. The FP-C:H films with superlubricity properties obtained under various conditions and the mature PECVD technology could promote the application of the superlubricity technology.Acknowledgements
The authors are grateful to the National Key Basic Research and Development (973) Program of China (Grant No. 2013CB632300), the National Natural Science Foundation of China (Grant No. 51275508, 51305434), and the CAS “Light of West China” Program.References
- A. Z. Szeri, Tribology: Friction, Lubrication, and Wear, Hemisphere, New York, 1980 Search PubMed.
- J. Li, C. Zhang, M. Deng and J. Luo, RSC Adv., 2015, 5, 30861–30868 RSC.
- R. Quadrelli and S. Peterson, Energy Policy, 2007, 35, 5938–5952 CrossRef.
- K. M. Liechti, Science, 2015, 348, 632–633 CrossRef CAS PubMed.
- J. Yuan and K. M. Liew, RSC Adv., 2014, 4, 45425–45432 RSC.
- O. Hod, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075444 CrossRef.
- M. Dienwiebel, G. S. Verhoeven, N. Pradeep, J. W. Frenken, J. A. Heimberg and H. W. Zandbergen, Phys. Rev. Lett., 2004, 92, 126101 CrossRef PubMed.
- Z. Liu, J. Yang, F. Grey, Z. Liu, Y. Liu, Y. Wang, Y. Yang, Y. Cheng and Q. Zheng, Phys. Rev. Lett., 2012, 108, 205503 CrossRef PubMed.
- K. Miura, S. Kamiya and N. Sasaki, Phys. Rev. Lett., 2003, 90, 055509 CrossRef CAS PubMed.
- A. Erdemir, O. L. Eryilmaz and G. Fenske, J. Vac. Sci. Technol., A, 2000, 18, 1987–1992 CAS.
- A. R. Konicek, D. S. Grierson, P. U. P. A. Gilbert, W. G. Sawyer, A. V. Sumant and R. W. Carpick, Phys. Rev. Lett., 2008, 100, 235502 CrossRef CAS PubMed.
- J. A. Ruan and B. Bhushan, J. Appl. Phys., 1994, 76, 8117–8120 CrossRef CAS.
- J. Xiao, L. Zhang, K. Zhou, J. Li, X. Xie and Z. Li, Carbon, 2013, 65, 53–62 CrossRef CAS.
- K. Miura, S. Kamiya and N. Sasaki, Phys. Rev. Lett., 2003, 90, 055509 CrossRef CAS PubMed.
- A. Smolyanitsky, RSC Adv., 2015, 5, 29179–29184 RSC.
- D. Berman, S. A. Deshmukh, S. K. Sankaranarayanan, A. Erdemir and A. V. Sumant, Science, 2015, 348, 1118–1122 CrossRef CAS PubMed.
- D. M. Mattox and V. H. Mattox, Vacuum coating technology, William Andrew Publishing, New York, 2003 Search PubMed.
- J. Cumings and A. Zettl, Science, 2000, 289, 602–604 CrossRef CAS PubMed.
- A. Kis, K. Jensen, S. Aloni, W. Mickelson and A. Zettl, Phys. Rev. Lett., 2006, 97, 025501 CrossRef CAS PubMed.
- Q. Zeng, O. Eryilmaz and A. Erdemir, RSC Adv., 2015, 5, 93147–93154 RSC.
- X. Liu, J. Yang, J. Hao, J. Zheng, Q. Gong and W. Liu, Adv. Mater., 2012, 24, 4614–4617 CrossRef CAS PubMed.
- B. Deepthi and H. C. Barshilia, Aerosp. Mater. Handb., 2012, p. 359 Search PubMed.
- Z. Wang, Z. Liu, M. A. Monne, S. Wang, Q. Yu and M. Y. Chen, RSC Adv., 2016, 6, 24865–24870 RSC.
- X. H. Lin and J. G. Gai, RSC Adv., 2016, 6, 17818–17844 RSC.
- G. Chen, Y. Seki, H. Kimura, S. Sakurai, M. Yumura, K. Hata and D. N. Futaba, Sci. Rep., 2014, 4, 3804 Search PubMed.
- N. M. Briggs and S. P. Crossley, RSC Adv., 2015, 5, 83945–83952 RSC.
- K. Teii, IEEE Trans. Plasma Sci., 2014, 42, 3862–3869 CrossRef CAS.
- G. N. Churilov, P. V. Novikov, V. E. Tarabanko, V. A. Lopatin, N. G. Vnukova and N. V. Bulina, Carbon, 2002, 40, 891–896 CrossRef CAS.
- O. S. Panwar, R. K. Tripathi, A. K. Srivastava, M. Kumar and S. Kumar, Diamond Relat. Mater., 2012, 25, 63–72 CrossRef CAS.
- O. S. Panwar, A. K. Srivastava, S. Kumar, R. K. Tripathi, M. Kumar and S. Singh, Surf. Coat. Technol., 2011, 206, 155–164 CrossRef.
- R. K. Tripathi, O. S. Panwar, A. K. Srivastava, I. Rawal and S. Chockalingam, Talanta, 2014, 125, 276–283 CrossRef CAS PubMed.
- O. S. Panwar, S. Kumar, A. K. Srivastava, A. Chouksey, R. K. Tripathi and A. Basu, Mater. Chem. Phys., 2012, 132, 659–666 CrossRef CAS.
- R. K. Tripathi, O. Panwar, A. K. Srivastava, M. Kumar and S. Chockalingam, J. Nanosci., 2013, 401710 Search PubMed.
- C. Wang, D. Diao, X. Fan and C. Chen, Appl. Phys. Lett., 2012, 100, 231909 CrossRef.
- E. H. T. Teo, J. Kulik, Y. Kauffmann, R. Kalish and Y. Lifshitz, Appl. Phys. Lett., 2011, 98, 123104 CrossRef.
- K. S. Subrahmanyam, L. S. Panchakarla, A. Govindaraj and C. N. R. Rao, J. Phys. Chem. C, 2009, 113, 4257–4259 CAS.
- O. S. Panwar, A. K. Kesarwani, S. R. Dhakate, B. P. Singh, R. K. Rakshit, A. Bisht and S. Chockalingam, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2013, 31, 040602 Search PubMed.
- A. K. Kesarwani, O. S. Panwar, S. Chockalingam, A. Bisht, S. R. Dhakate, B. P. Singh and R. K. Rakshit, Sci. Adv. Mater., 2014, 6, 2124–2133 CrossRef CAS.
- A. K. Kesarwani, O. S. Panwar, S. R. Dhakate, R. K. Rakshit, V. N. Singh, A. Bisht and A. Kumar, J. Vac. Sci. Technol., A, 2016, 34, 021504 Search PubMed.
- A. Bisht, S. Chockalingam, O. S. Panwar, A. K. Kesarwani, B. P. Singh and V. N. Singh, Sci. Adv. Mater., 2015, 7, 1424–1434 CrossRef CAS.
- S. Chockalingam, A. Bisht, O. S. Panwar, A. K. Kesarwani, B. P. Singh, J. Chand and V. N. Singh, Mater. Chem. Phys., 2015, 156, 38–46 CrossRef CAS.
- V. Kouznetsov, K. Macak, J. M Schneider, U. Helmersson and I. Petrov, Surf. Coat. Technol., 1999, 122, 290–293 CrossRef CAS.
- D. Lundin and H. Pedersen, Phys. Procedia, 2013, 46, 3–11 CrossRef CAS.
- Y. Wang, D. C. Alsmeyer and R. L. McCreery, Chem. Mater., 1990, 2, 557–563 CrossRef CAS.
- S. N. Magonov, V. Elings and M. H. Whangbo, Surf. Sci., 1997, 375, 385–391 CrossRef.
- L. G. Cançado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, N. L. Speziali, A. Jorio and M. A. Pimenta, Carbon, 2008, 46, 272–275 CrossRef.
- M. S. Dresselhaus, A. Jorio, A. G. Souza Filho and R. Saito, Philos. Trans. R. Soc., A, 2010, 368, 5355–5377 CrossRef CAS PubMed.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. R. P. Silva, J. Appl. Phys., 1996, 80, 440–447 CrossRef CAS.
- H. Ehrhardt, R. Kleber, A. Kru, W. Dworschak, K. Jung, I. Mu and H. Metz, Diamond Relat. Mater., 1992, 1, 316–320 CrossRef CAS.
- R. Kleber, K. Jung, H. Ehrhardt, I. Mühling, K. Breuer, H. Metz and F. Engelke, Thin Solid Films, 1991, 205, 274–278 CrossRef CAS.
- M. Mowry, D. Palaniuk, C. C. Luhrs and S. Osswald, RSC Adv., 2013, 3, 21763–21775 RSC.
- E. M. Ferreira, M. V. Moutinho, F. Stavale, M. M. Lucchese, R. B. Capaz, C. A. Achete and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125429 CrossRef.
- L. M. Malard, M. H. D. Guimaraes, D. L. Mafra and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125426 CrossRef.
- C. W. Huang, B. J. Lin, S. Y. Juang, F. Y. Shih, W. H. Wang, C. Y. Liu and H. C. Chui, RSC Adv., 2014, 4, 51067–51071 RSC.
- Q. Wu, Y. Shen, Q. Wang, F. Gu, M. Cao and L. Wang, in Eighth International Conference on Thin Film Physics and Applications, 2013, 90681T-90681T Search PubMed.
- J. Filik, P. W. May, S. R. J. Pearce, R. K. Wild and K. R. Hallam, Diamond Relat. Mater., 2003, 12, 974–978 CrossRef CAS.
- J. L. Hueso, J. P. Espinós, A. Caballero, J. Cotrino and A. R. González-Elipe, Carbon, 2007, 45, 89–96 CrossRef CAS.
- W. H. Lee, J. G. Lee and P. J. Reucroft, Appl. Surf. Sci., 2001, 171, 136–142 CrossRef CAS.
- O. L. Eryilmaz and A. Erdemir, Tribol. Lett., 2007, 28, 241–249 CrossRef CAS.
- A. H. Lettington, Carbon, 1998, 36, 555–560 CrossRef CAS.
- A. Erdemir, J. Fontaine and C. Donnet, Tribology of Diamond-Like Carbon Films, Springer, US, 2008, pp. 237–262 Search PubMed.
- C. A. Freyman, Y. F. Chen and Y. W. Chung, Surf. Coat. Technol., 2006, 201, 164–167 CrossRef CAS.
- A. Chuvilin, U. Kaiser, E. Bichoutskaia, N. A. Besley and A. N. Khlobystov, Nat. Chem., 2010, 2, 450–453 CrossRef CAS PubMed.
- Y. E. Lozovik and A. M. Popov, Phys.–Usp., 1997, 167, 751–774 CAS.
- J. Y. Huang, F. Ding, K. Jiao and B. I. Yakobson, Phys. Rev. Lett., 2007, 99, 175503 CrossRef CAS PubMed.
- L. Pauling, The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry, Cornell university press, 1960 Search PubMed.
- L. Kong, A. Enders, T. S. Rahman and P. A. Dowben, J. Phys.: Condens. Matter, 2014, 26, 443001 CrossRef PubMed.
- H. Ulbricht, R. Zacharia, N. Cindir and T. Hertel, Carbon, 2006, 44, 2931–2942 CrossRef CAS.
- O. M. Braun, Phys. Rev. Lett., 2005, 95, 126104 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |