
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 13N-labelled polysubstituted triazoles via Huisgen cycloaddition†
S. M.
Joshi
a,
V.
Gómez-Vallejo
b,
V.
Salinas
b and
J.
Llop
*a
aRadiochemistry and Nuclear Imaging Group, CIC biomaGUNE, Paseo Miramón 182, Parque Tecnológico de San Sebastián, 20009 Donostia-San Sebastián, Gipuzkoa, Spain. E-mail: jllop@cicbiomagune.es
bRadiochemistry Platform, CIC biomaGUNE, Paseo Miramón 182, Parque Tecnológico de San Sebastián, 20009 Donostia-San Sebastián, Gipuzkoa, Spain
First published on 8th November 2016
Abstract
The use of the positron emitter nitrogen-13 (13N) has been historically restricted due to its short half-life (T1/2 = 9.97 min). However, its stable isotopes (nitrogen-14 and nitrogen-15) are present in many biologically active molecules; therefore, the incorporation of 13N in the toolbox of PET chemists might be a valuable option for the preparation of new labelled compounds or incorporation of the label in different positions. Here we present the unprecedented radiosynthesis of 13N-labelled polysubstituted triazoles via Huisgen cycloaddition by reaction of 13N-labelled aromatic azides with alkyne derivatives and aldehydes. Six different 13N-labelled triazoles were successfully synthesized. After automatization of the synthetic process and optimization of experimental conditions, one selected triazole could be prepared with high radiochemical purity and decay-corrected radiochemical yields of 11 ± 2%. The amount of activity obtained should be sufficient to approach future in vitro and in vivo studies. The novel methodology might open new avenues for the preparation of radiotracers which cannot be labelled using other more conventional positron emitters.
Introduction
Positron Emission Tomography (PET) is a minimally invasive molecular imaging technique that enables the determination of the spatiotemporal distribution of a radiolabelled molecule (radiotracer) after administration to a living organism. Due to the high sensitivity of PET, the radiotracer is usually administered in the sub-micromolar range, facilitating the investigation of biological or physiological processes in vivo without having any toxicological, pharmacological and/or undesired side effects.Among all positron emitters, fluorine-18 (18F) and carbon-11 (11C) have been the most widely used.1 Fluorine-18 has a relatively long half-life (T1/2 = 109.7 min), facilitating the centralised production of radiotracers and distribution to surrounding centres. It decays almost quantitatively by positron emission and has a short positron range (Eβmax = 0.64 MeV, maximum range in water = 2.4 mm), which ultimately results in higher resolution images. Carbon-11 has a shorter half-life (T1/2 = 20.4 min) and a longer positron range (Eβmax = 0.96 MeV, maximum range in water = 3.8 mm); however, it can be produced in different chemical forms in biomedical cyclotrons; additionally, because all organic molecules contain stable carbon atoms in their structure, the use of carbon-11 enables the preparation of radiotracers identical to the non radioactive molecule to be investigated.
The use of shorter half-lived positron emitters such as nitrogen-13 (13N, T1/2 = 9.97 min, Eβmax = 1.19 MeV, maximum range in water = 5.4 mm) has been historically much more restricted, and only a few studies describing new synthetic strategies for the incorporation of this radioisotope into bioactive molecules have been reported to date.2 However, 13N can be efficiently produced in different chemical forms in biomedical cyclotrons (e.g. [13N]NH4+, [13N]N2, [13N]NO3−) by proton irradiation of natural oxygen via the 16O(p,α)13N nuclear reaction. Additionally, its stable isotopes (nitrogen-14 and nitrogen-15) are present in the majority of biological active molecules. Consequently, the incorporation of 13N in the toolbox of PET chemists might be a valuable alternative for the preparation of new labelled compounds or incorporation of the label in different positions, which may provide complementary information regarding the in vivo behaviour and metabolism of the radiotracers.
During the last decade, we have developed strategies for the preparation of 13N-labelled amino acids using enzymatic reactions and [13N]NH3 as the labelling agent,3 as well as the chemical4 and biocatalysed5 reduction of [13N]NO3− into [13N]NO2− and subsequent preparation of [13N]nitrosamines,6 [13N]nitrosothiols4,7 and [13N]azo derivatives.8,9 Very recently, we investigated the preparation of 13N-labelled phenyl azides by reaction of an aromatic amine with [13N]NO2− and hydrazine hydrate to demonstrate that the formation of aryl azides from the corresponding diazonium salts occurs via a stepwise mechanism via acyclic zwitterionic intermediates. These results were confirmed by theoretical calculations.10
In continuation of our work, we present here the unprecedented radiosynthesis of 13N-labelled polysubstituted triazoles via Huisgen cycloaddition using [13N]NO2− as the primary labelling agent. The 4-step process consisted of: (i) chemical reduction of [13N]NO3− into [13N]NO2−; (ii) formation of the 13N-labelled diazonium salts by reaction of [13N]NO2− with aromatic amines under acidic conditions; (iii) formation of the 13N-labelled azides by reaction of the 13N-labelled diazonium salts with NaNO2 and hydrazine hydrate in acidic media; and (iv) reaction of the 13N-labelled azide with alkynes or aldehydes in the presence of a catalyst (see Fig. 1). After optimization of the experimental conditions and automatisation of the whole process, one selected 13N-labelled triazole could be obtained in acceptable non decay-corrected radiochemical yields, which can be anticipated to suffice for the approach of future in vitro or in vivo studies. The methodology developed here could be applied to the preparation of a wide range of substituted triazoles, and may become relevant in those cases in which the radiolabelling process using more conventional positron emitters (i.e.18F or 11C) is challenging or not viable.
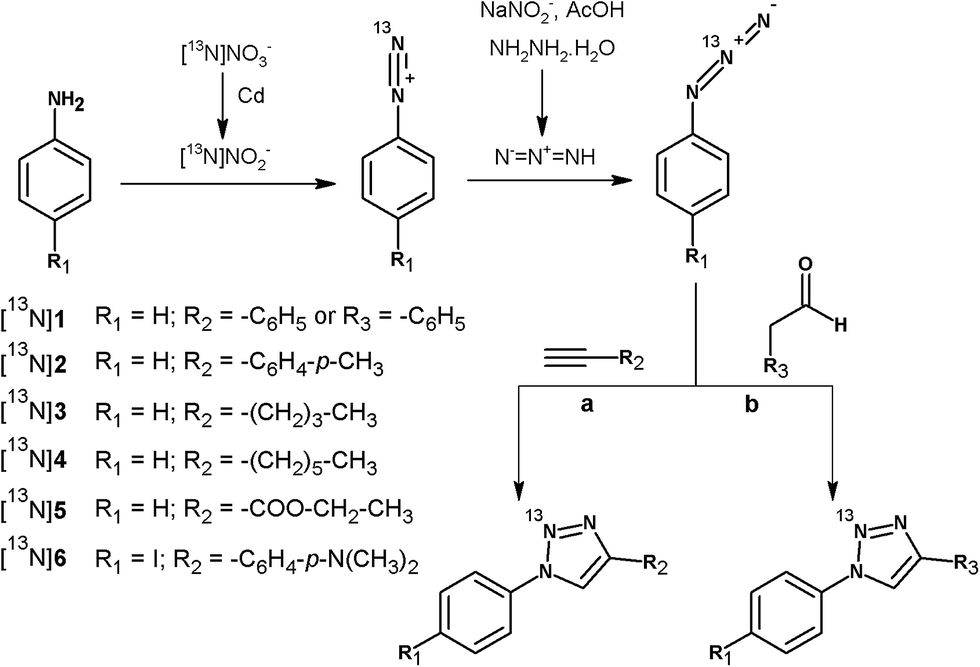 | ||
| Fig. 1 Scheme of the 4-step synthetic process followed for the preparation of 13N-labelled substituted triazoles by reaction of 13N-labelled azides with alkynes (a) and aldehydes (b). | ||
Experimental
General information
Aniline (reagent plus grade 99%), 4-iodoaniline (98%), sodium nitrite (ACS reagent, 97%), acetic acid (Reagentplus®, >99%), hydrazine hydrate solution (iodometric, 78–82%), cyclohexylamine (Reagentplus®, >99%), paraformaldehyde (reagent plus grade crystalline), glyoxal solution (40 wt% in H2O), sodium carbonate (ACS reagent, anhydrous), magnesium sulphate (ACS reagent, anhydrous, >99.5%), sodium sulphate (ACS reagent, anhydrous, >99%), tetrakis(acetonitrile)copper(I) hexafluorophosphate (97%), sodium tert-butoxide (97%), Celite® S (filter aid, dried, untreated), copper(II) sulphate pentahydrate (ACS reagent, >98%), sodium ascorbate (Pharmaceutical secondary standard), 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU, 98%), azidobenzene solution (0.5 M in tert-butyl methyl ether, >95%), 1-azido-4-iodobenzene solution (0.5 M in tert-butyl methyl ether, >95%), 4-ethynyl-N,N-dimethylaniline (97%), phenyl acetylene (98%), ethyl propiolate (99%), 1-hexyne (97%), 1-octyne (97%), 4-ethynyltoluene (97%), phenylacetaldehyde (>90%), hydrochloric acid (37%, extrapure, Ph. Eur.), tetrahydrofuran (absolute over molecular sieve, >99.5%), and dimethyl sulfoxide (anhydrous, >99.9%) were purchased from Sigma-Aldrich and used without further purification. Hexane (synthesis grade), ethyl acetate (synthesis grade), dichloromethane (synthesis grade), acetonitrile (HPLC grade) and diethyl ether (extra pure) were purchased from Scharlab (Sentmenat, Barcelona, Spain). Ultrapure water (Type I water, ISO 3696) was obtained from a Milli-Q® purification system (Merck Millipore).Synthetic procedures: chemistry
Synthetic procedures: radiochemistry
Results and discussion
Nitrogen-13 can be produced in different chemical forms in biomedical cyclotrons and offers a wide variety of synthetic possibilities for the preparation of PET radiotracers. However, its short half life demands for the development of fast, efficient and robust synthetic processes. This was especially relevant in the work reported here, because the synthetic strategy was envisioned through a 4-steps process plus one intermediate and one final purification steps.In our previous work,10 we demonstrated that two approaches could be used for the synthesis of 13N-labelled aryl azides: (i) reaction of aniline with sodium nitrite to yield the non-labeled diazonium salt and subsequent reaction with 13N-labelled azide ion (prepared by reaction of hydrazine hydrate with [13N]NO2− in acidic media); and (ii) reaction of aniline with [13N]NO2− to yield the 13N-labelled diazonium salt and subsequent reaction with azide ion (prepared by reaction of hydrazine hydrate with sodium nitrite in acidic media). Our results showed that the radiolabel was almost quantitatively transferred to the azide under route (ii). Consequently, this route was considered as the most appropriate to achieve optimal results and the experimental conditions of this part of the reaction were not further optimised.
It is worth mentioning that during the preparation of labelled azides it is paramount to work under no-carried-added conditions, in order to achieve high specific activity (amount of radioactivity per unit mass) values in the final labelled triazoles. In other words, the preparation of the labelled diazonium salt had to be conducted without addition of non-radioactive NO2−. This, ultimately, led to the situation in which the labelled specie in the (3 + 2) cycloaddition reaction for the formation of the 13N-labelled triazoles is the limiting reagent, as its concentration is much lower than any other reagent. Taking this into account, we first tackled the optimization of the experimental conditions for the preparation of 13N-labelled triazoles by reaction of the labelled azides with alkynes (see Fig. 1). During this process, the work was conducted manually to have a better control of the individual steps. In all cases, control analysis performed after the reduction step showed that 70–80% of the radioactivity eluted from the cadmium column was due to [13N]NO2−; [13N]NH4+ and non-reduced [13N]NO3− accounted for around 2% and 18–28% of the total radioactivity, respectively. These results are in good agreement with previous data.6–9,15
After formation of the azide, substantial amount of chemical impurities plus [13N]NH4+, [13N]NO3− and unreacted [13N]NO2− were present in the reaction mixture. Additionally, subsequent reaction for the formation of the labelled triazole should be conducted in non-aqueous media. Because of this, the implementation of an intermediate step to change the solvent was required and we first considered liquid–liquid extraction with dichloromethane. Radio-HPLC analysis of both the aqueous and organic phases confirmed quantitative extraction of the labelled azide: no peak corresponding to this specie was found in the aqueous phase. Additionally, no radiochemical impurities (i.e. [13N]NH4+, [13N]NO2−, [13N]NO3− or labelled diazonium salt) were found in the organic phase, although the presence of the starting aniline could be detected. This impurity was anticipated not to interfere in the following catalytic reaction.
To conduct the (3 + 2) cycloaddition under hot conditions, our first attempts were performed using CuSO4·H2O as the catalyst, mimicking the reaction conditions used during preparation of the non-radioactive analogues. With that aim, the preparation of [13N]1 was approached by reaction of [13N]phenylazide (redissolved in acetonitrile after evaporation of dichloromethane) with phenylacetylene in the presence of the catalyst (2 mg) for 10 min at RT. Unfortunately, the formation of the desired labelled triazole could not be detected by radio-HPLC. Similar results were obtained with copper(II) oxide (CuO) and copper(I) iodide as the catalyst. In view of these results, we decided to use [(Icy)2Cu]PF6, which has proven efficient in a wide variety of (3 + 2) cycloaddition reactions.14 In this case, the reaction at RT for 10 minutes offered radiochemical conversion values (RCC, calculated from radio-chromatographic profiles, expressed as the ratio between the area under the peak corresponding to [13N]triazole and the sum of the areas for all the peaks in the chromatogram, in percentage) of 15 ± 2% (Table 1, entry 1).
| Entry | Triazole | RT | 50a °C/60b °C |
|---|---|---|---|
| a Reaction of azide with alkyne. b Reaction of azide with aldehyde; values are expressed as average ± standard deviation, n = 3; RT: room temperature; reaction time was 10 min in all cases. | |||
| 1 | [13N]1a | 15 ± 2 | 49 ± 8 |
| 2 | [13N]2a | 13 ± 3 | 42 ± 4 |
| 3 | [13N]3a | 38 ± 14 | 92 ± 8 |
| 4 | [13N]4a | 10 ± 4 | 70 ± 8 |
| 5 | [13N]5a | 72 ± 3 | 94 ± 6 |
| 6 | [13N]6a | 17 ± 6 | 38 ± 2 |
| 7 | [13N]1b | 23 ± 5 | 96 ± 2 |
After these encouraging results, we decided to extend the methodology to the preparation of other functionalized triazoles ([13N]2–[13N]6), and different reaction temperatures were assayed. As it can be seen in Table 1, our method enabled the preparation of all labelled triazoles. Radiochemical conversion values increased with temperature to reach acceptable values (38–94%) when the reaction was carried out for 10 minutes at 50 °C (Table 1, entries 1–6; see Fig. 2a and b for examples of chromatographic profiles corresponding to the synthesis of [13N]1 at 50 °C).
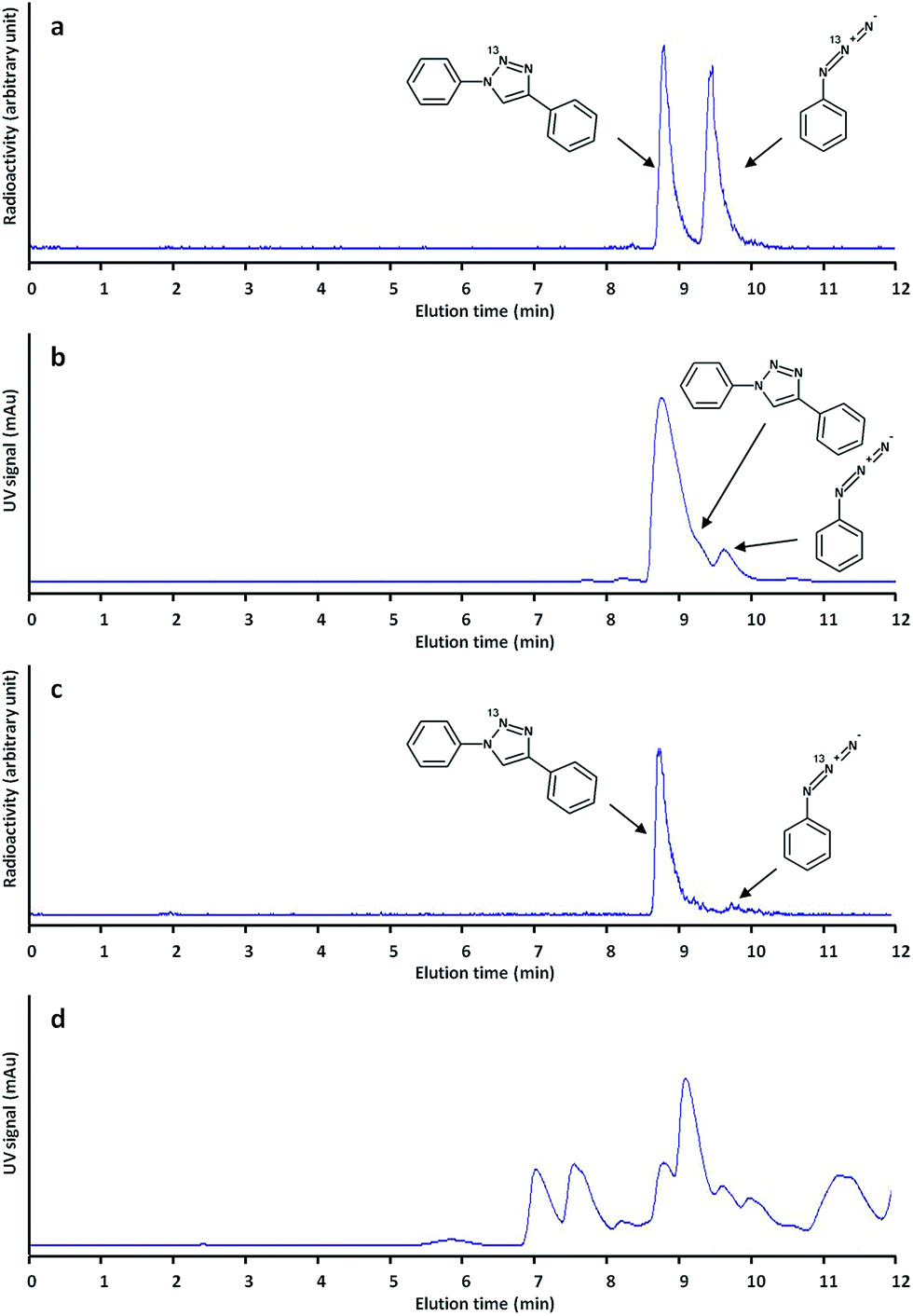 | ||
| Fig. 2 Chromatographic profiles using radioactive (a and c) and UV (b and d) detection after analysis of the reaction crude corresponding to the synthesis of [13N]1; (a and b) synthesis by (3 + 2) cycloaddition using labelled phenylazide with phenylacetylene; (c and d) synthesis by (3 + 2) cycloaddition using labelled phenylazide with phenylacetaldehyde. | ||
Interestingly, high conversion values were obtained when aliphatic alkynes were used. This result is not in agreement with previous works reported in the literature, as aliphatic alkynes are known for their lower reactivity when compared to aromatic ones.16,17 However, long reaction times (hours) were used in these previous works. Additionally, because we conducted the reactions under no-carrier-added conditions, the 13N-labelled azide is the limiting reagent and its concentration is extremely low when compared to that of the alkyne. Despite further investigation would be required, we hypothesize that the low RCC values obtained with aromatic azides might be due to the kinetics of the reaction. Longer reaction times may lead to quantitative conversion, as observed in the case of aliphatic triazoles.
In view of the promising results, we decided to expand the scope of our work by tackling the preparation of [13N]1 by reaction of the labelled azide with phenylacetaldehyde. In our first attempts, [(Icy)2Cu]PF6 was used as the catalyst, but the formation of [13N]1 could not be detected by radio-HPLC. The formation of substituted triazoles by reaction of azides with aldehydes has been previously reported in the literature.18 In this previous work, 1,8-diazabicycloundec-7-ene (DBU) offered excellent results when the reaction was carried out in DMSO. In our hands, this catalyst offered good radiochemical conversion values (23 ± 5%, Table 1, entry 7) when the reaction was conducted in DMSO at 25 °C. The reaction was almost quantitative at T = 60 °C (see Fig. 2c and d for examples of chromatographic profiles).
Liquid–liquid extraction as an intermediate purification step is a very well established procedure for reactions that are conducted under non-radioactive conditions. However, such experimental step is sub-optimal in radioactive conditions because automatisation is extremely challenging. Hence, and moving towards the development of a fully automated process, other alternatives were explored. In previous works, our group has used solid phase extraction cartridges as a suitable tool to switch from aqueous to organic solvent during the synthesis of 13N-labelled azo compounds.7 Here, we anticipated that dilution of the reaction crude with water after formation of the labelled azide and subsequent elution through a C-18 cartridge would result in quantitative trapping of the labelled azide. However, first attempts performed with [13N]phenylazide were unsuccessful, and no trapping could be observed. Because the reaction for the formation of the 13N-labelled azide is carried out under strong acidic conditions, we postulated that the lack of retention might be due to the formation of the protonated form (positively charged) of the azide, which has low affinity for the C-18 phase. As expected, replacement of water by sodium acetate solution and elution through the C-18 cartridge resulted in quantitative trapping of the labelled azide, as revealed by radio-HPLC analysis of the eluate. In the case of [13N]p-iodophenylazide, used in the preparation of compound [13N]6, dilution with water led to quantitative trapping, probably due to the presence of the iodine atom, which confers a stronger hydrophobic character to the molecule, regardless of the net charge.
In all cases, flushing of the C-18 cartridge with helium gas for 1 min was sufficient to remove the majority of the water; subsequent elution of the trapped labelled species with acetonitrile and reaction with the corresponding alkyne under catalytic conditions led to the formation of the desired labelled compounds ([13N]1–[13N]6) with RCC values equivalent to those shown in Table 1.
Incorporation of the solid phase extraction process as an intermediate purification step enabled the automatisation of the whole process (including final purification by semi-preparative HPLC) using the Tracerlab™ FXFE synthesis module, with appropriate adaptation (see Fig. S1 and ESI† for further experimental details). The experimental conditions for the preparation of compound [13N]6 were further optimized at this step. This triazole was selected for full automatisation because its non radioactive analogue has shown promising properties to target β-amyloid aggregates, and hence it may find application in the early diagnose or evaluation of response to treatment in Alzheimer's disease.12 The amount of catalyst, the reaction temperature and the reaction time for the formation of the labelled triazole were varied (Table 2).
| Entry | T (°C) | t (min) | Catalyst (mg) | RCC (%) |
|---|---|---|---|---|
| a Values are expressed as average ± standard deviation, n = 3; catalyst: [(Icy)2Cu]PF6; RCC: radiochemical conversion. | ||||
| 1 | 50 | 2 | 2 | 0 |
| 2 | 50 | 2 | 5 | 0 |
| 3 | 50 | 2 | 10 | 0 |
| 4 | 50 | 5 | 2 | 16 ± 5 |
| 5 | 50 | 5 | 5 | 20 ± 4 |
| 6 | 50 | 5 | 10 | 22 ± 5 |
| 7 | 50 | 10 | 2 | 36 ± 4 |
| 8 | 50 | 10 | 5 | 49 ± 7 |
| 9 | 50 | 10 | 10 | 75 ± 8 |
| 10 | 80 | 5 | 10 | 40 ± 3 |
| 11 | 80 | 10 | 10 | 83 ± 6 |
| 12 | 110 | 5 | 10 | 85 ± 9 |
| 13 | 110 | 10 | 10 | 96 ± 3 |
As it can be seen in the table, no reaction was observed when the reaction time was set to 2 min, irrespective of the amount of catalyst (entries 1–3). It is worth mentioning that heating of the vial where the reaction is carried out starts at t = 0. Hence, 2 min might not be sufficient to reach the desired temperature in the reaction mixture. In general terms, higher temperature, longer reaction times and higher amount of catalyst led to higher RCC values, as expected. At optimal reaction temperature (110 °C) and amount of catalyst (10 mg), the reaction time of 10 min proved to be less desirable than the 5 min period, because the gain in RCC (approximately 10%, from 85 to 96%, see entries 12 and 13 in Table 2) was negligible compared with the decay of nitrogen-13 during the extra 5 min gap (close to 50%). Hence, T = 110 °C, t = 5 min and amount of catalyst = 10 mg were established as optimal conditions among the investigated scenarios.
These experimental conditions were applied for the fully automated preparation of pure [13N]6 (see ESI† for experimental details). After purification by semi-preparative HPLC (see Fig. 3a for example of chromatographic profile) isolated radiochemical yields of 11 ± 2%, related to the starting amount of 13N produced in the cyclotron and decay corrected to the end of the irradiation process, were obtained in overall production times of 25 min (non-decay corrected radiochemical yield = 2 ± 0.4%). Specific activities at the end of the synthesis were 4.6 ± 0.2 GBq μmol−1, and radiochemical purity was >99% in all cases, as determined by radio-HPLC. No chemical impurities were detected by HPLC (Fig. 3c). Despite non-decay corrected radiochemical yields are low, an irradiation of 4 μA h yields around 6.6 GBq of 13N in the cyclotron target. Hence, around 130 MBq (3.6 mCi) of pure radiotracer could be obtained at the end of the preparation process. Typical injected doses in mice and rats are around 100 and 500 μCi, respectively. Hence, the here reported methodology should enable subsequent in vitro or in vivo studies in small animal species.
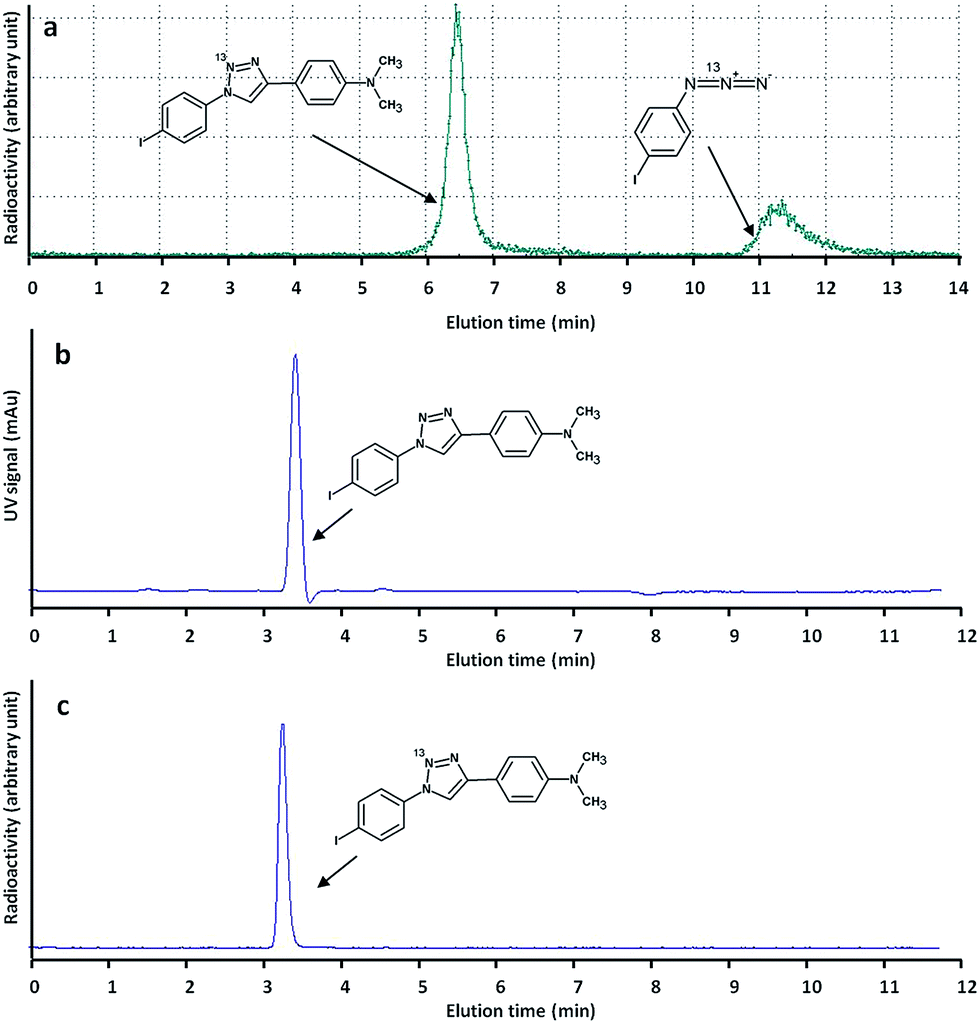 | ||
| Fig. 3 (a) Chromatographic profile (radioactive detector) corresponding to the purification of [13N]6. Two radioactive peaks, corresponding to [13N]6 and unreacted 13N-labelled 1-azido-4-iodobenzene, were found; (b and c) chromatographic profiles (UV-Vis and radioactive detectors, respectively) corresponding to the analysis of purified [13N]6. No chemical nor radiochemical impurities were detected. | ||
Conclusions
In conclusion, we present here an unprecedented and fully automated methodology for the preparation of chemically and radiochemically pure 13N-labelled substituted triazoles by reaction of 13N-labelled azides with alkynes. By accurate selection of the catalyst, the methodology could be extended to the preparation of triazoles using aldehydes instead of alkynes in the (3 + 2) cycloaddition step. Sufficient amount of radiotracer to approach subsequent in vitro and in vivo studies in small animal species could be obtained after optimization of the experimental conditions. Radiolabelling with 13N may find interesting applications, especially in those occasions in which incorporation of other positron emitters into the target molecule is not feasible, or to tackle the incorporation of the label in different positions to enable accurate metabolic studies.Acknowledgements
All the work has been supported by the RADIOMI project (EU FP7-PEOPLE-2012-ITN/316882).Notes and references
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- V. Gómez-Vallejo, V. Gaja, K. B. Gona and J. Llop, J. Labelled Compd. Radiopharm., 2014, 57, 244–254 CrossRef PubMed.
- E. S. da Silva, V. Gómez-Vallejo, Z. Baz, J. Llop and F. López-Gallego, Chem.–Eur. J., 2016, 22, 13619–13626 CrossRef CAS PubMed.
- J. Llop, V. Gómez-Vallejo, M. Bosque, G. Quincoces and I. Peñuelas, Appl. Radiat. Isot., 2009, 67, 95–99 CrossRef CAS PubMed.
- E. S. Da Silva, V. Gómez-Vallejo, J. Llop and F. López-Gallego, Catal.: Sci. Technol., 2015, 5, 2705–2713 CAS.
- V. Gómez-Vallejo, K. Kato, M. Hanyu, K. Minegishi, J. I. Borrell and J. Llop, Bioorg. Med. Chem. Lett., 2009, 19, 1913–1915 CrossRef PubMed.
- V. Gómez-Vallejo, K. Kato, I. Oliden, J. Calvo, Z. Baz, J. I. Borrell and J. Llop, Tetrahedron Lett., 2010, 51, 2990–2993 CrossRef.
- V. Gaja, V. Gómez-Vallejo, M. Puigivila, C. Pérez-Campaña, A. Martin, A. García-Osta, T. Calvo-Fernández, M. Cuadrado-Tejedor, R. Franco and J. Llop, Mol. Imaging Biol., 2014, 16, 538–549 CrossRef PubMed.
- V. Gómez-Vallejo, J. I. Borrell and J. Llop, Eur. J. Med. Chem., 2010, 45, 5318–5323 CrossRef PubMed.
- S. M. Joshi, A. De Cózar, V. Gómez-Vallejo, J. Koziorowski, J. Llop and F. P. Cossío, Chem. Commun., 2015, 51, 8954–8957 RSC.
- A. D. Moorhouse and J. E. Moses, Synlett, 2008, 2089–2092, DOI:10.1055/s-2008-1078019.
- W. Qu, M. P. Kung, C. Hou, S. Oya and H. F. Kung, J. Med. Chem., 2007, 50, 3380–3387 CrossRef CAS PubMed.
- B. Kaboudin, R. Mostafalu and T. Yokomatsu, Green Chem., 2013, 15, 2266–2274 RSC.
- S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2008, 47, 8881–8884 CrossRef PubMed.
- V. Gaja, V. Gómez-Vallejo, M. Cuadrado-Tejedor, J. I. Borrell and J. Llop, J. Labelled Compd. Radiopharm., 2012, 55, 332–338 CrossRef CAS.
- A. Krasiński, V. V. Fokin and K. B. Sharpless, Org. Lett., 2004, 6, 1237–1240 CrossRef PubMed.
- X. Meng, X. Xu, T. Gao and B. Chen, Eur. J. Org. Chem., 2010, 5409–5414, DOI:10.1002/ejoc.201000610.
- D. B. Ramachary, A. B. Shashank and S. Karthik, Angew. Chem., Int. Ed., 2014, 53, 10420–10424 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Design and experimental details for automatic radiosynthesis; structural characterization of the reference compounds. See DOI: 10.1039/c6ra24670b |
| This journal is © The Royal Society of Chemistry 2016 |