
Synthesis of core–shell imprinting polymers with uniform thin imprinting layer via iniferter-induced radical polymerization for the selective recognition of thymopentin in aqueous solution
 *a
Chunbao
Du,
a
*a
Chunbao
Du,
a
Abstract
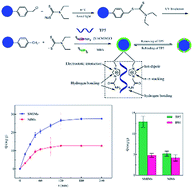
An approach for synthesizing core–shell imprinting polymers using P(EGDMA-CMS) microspheres prepared via dispersion polymerization as a core and employing a surface imprinting technique and iniferter-induced radical polymerization is described. N,N-Diethyldithiocarbamyl groups were immobilized on the surface of the supporting microspheres to form the surface iniferter and further prepare the imprinting layer. Thymopentin (TP5) was selected as the template molecule, which was known to be an immunomodulating agent that had medical properties. Here, a bifunctional ionic liquid (IL), namely, 1-vinyl-3-carbamoylmethyl-imidazolium chloride ([VACMIM]Cl), was synthesized and employed as a novel functional monomer on the basis of the demands of peptide imprinting and the designability of ILs. Under irradiation by UV light, the surface iniferter decomposed and then polymerization was initiated to form a thin surface imprinting layer with specific recognition cavities for TP5. The surface imprinting layer possessed a uniform thickness of ∼35 nm, which was beneficial for the mass transfer of the template TP5, owing to good control of the thickness of the imprinting layer by controlled/living radical polymerization (CRP). The polymeric microspheres were fully characterized and their adsorption properties were investigated. The surface molecular imprinting microspheres (SMIMs) displayed high binding affinity, good selective specificity, rapid adsorption equilibrium and satisfactory reusability. The Scatchard plots of the SMIMs could be fitted to one straight line, which suggested that there was only one kind of binding site. Furthermore, the method of combining a surface imprinting technique and CRP together can be extended to a wide range of applications for chemical sensors, drug delivery and the separation of biomacromolecules.
 Please wait while we load your content...
Please wait while we load your content...

