
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailored activated carbons for supercapacitors derived from hydrothermally carbonized sugars by chemical activation†
Kian Keat Lee a,
Wenming Haoa,
Mikaela Gustafssona,
Cheuk-Wai Taia,
Daniel Morinb,
Eva Björkmanc,
Malte Lilliestrålec,
Fredrik Björeforsd,
Anna M. Andersson*b and
Niklas Hedin*a
a,
Wenming Haoa,
Mikaela Gustafssona,
Cheuk-Wai Taia,
Daniel Morinb,
Eva Björkmanc,
Malte Lilliestrålec,
Fredrik Björeforsd,
Anna M. Andersson*b and
Niklas Hedin*a
aDepartment of Materials and Environmental Chemistry-Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: niklas.hedin@mmk.su.se
bABB Corporate Research, Forskargränd 7, SE-721 78 Västerås, Sweden. E-mail: anna.m.andersson@se.abb.com
cBiokol Lilliestråle & Co KB, Sibyllegatan 53, SE-114 43 Stockholm, Sweden
dDepartment of Chemistry-Ångström Laboratory, Uppsala University, Box 538, SE-751 21 Uppsala, Sweden
First published on 14th November 2016
Abstract
Activated carbons (ACs) are actively researched as electrode materials for supercapacitors and there is a significant interest to produce ACs from sustainable and low cost precursors. In this study, various ACs were prepared from hydrothermally carbonized sugars by KOH activation. Both the hydrothermal carbonization and activation processes were optimized to tailor the properties (e.g. textural properties, chemical composition, N-doping, electrical conductivity) of the ACs. For instance, the Brunauer–Emmett–Teller (BET) surface areas (SBET) were tuned in the range of 800–3000 m2 g−1 with associated variation in the extent of microporosity and pore size distributions (PSDs). The ACs were evaluated electrochemically as materials for supercapacitor electrodes in a symmetrical two-electrode cell using an aqueous electrolyte. The relationship between the electrochemical, textural, electrical, and physicochemical properties were analyzed systematically to understand the key factors determining the electrochemical performance. A high specific capacitance (Cm) of ∼260 F g−1 was achieved at a moderately high SBET of ∼1300 m2 g−1, which was equivalent to a Cm/SBET of 20 μF cm−2, for an optimal AC prepared from hydrothermally carbonized glucose. The very high surface-specific capacitance highlights that the specific surface area is certainly not the sole limiting parameter for effective electrode materials.
1. Introduction
Supercapacitors (or ultracapacitors) are electrochemical energy storage devices that store electricity electrostatically or electrochemically via redox reactions and electrosorption. Hence, they are closely related to batteries.1–4 With their high energy densities of >10 kW h kg, and discharge times that are in the range of milliseconds to minutes, many applications in the previously void region between conventional capacitors and batteries are enabled. For electrified vehicles, applications could be in regenerative braking systems, start–stop systems, and in power boosts for various driver interfaces such as facilitated steering or air conditioning.4–6 Since batteries and supercapacitors in many cases are complimentary, hybrid systems of both technologies are often judged to be attractive.2,5 The hybrid systems are suggested for large-scale energy storage to enable effective power compensation in electrical grids. Such a combined storage features short and long time scale compensation and is potentially very beneficial from a cost perspective.5Highly porous carbons are the primary choice of electrode materials for supercapacitors. This preference relates to their high specific surface area or high (ultra)micropore volume, which is necessary for a high electric double layer (EDL) capacitance; to their relatively high electrical conductivity, which is important for the power delivery; and to their low production cost, which is critical for industrial applications.1 By rationally tuning the textural, structural, and surface properties of carbon materials, optimum electrochemical performance can be achieved and adapted based on system requirements. Considering sustainability and economy, it is preferred that ACs are produced from renewable, sustainable, abundant and cost-effective resources. Carbohydrate-based ACs fit these demands as they can be manufactured reproducibly at scale from consistent and abundant sources. Hence, they are up-and-coming alternatives.7–10
Hydrothermal carbonization of biomasses and sugars produce hydrothermal carbons, at low temperatures (150–250 °C) and autogenous pressure.11 These hydrothermal carbons are suitable precursors to ACs. Compared with precursors produced by conventional pyrolysis, the energy cost of producing hydrothermal carbons is lower. This difference relate to the lower temperature, exothermic reactions, and no need for precursor drying.11–13 Moreover, the hydrothermal carbonization process is capable of homogenizing biomass precursors, removing certain mineral impurities, and providing an excellent carbon yield.14 The hydrothermal carbons often display uniform physico-chemical properties and various tunable functional groups (e.g. oxygen, nitrogen, phosphorus) that depend on the composition of the biomass, additives and the process conditions.15–18
As a simple form of hydrothermal carbon, hydrothermally carbonized glucose (HTC-glucose), has been increasingly studied since the early 2000s.11,19,20 The microspheres of HTC-glucose generally possess almost no open porosity, and post-heat treatment only leads to a moderately increased surface area (400 m2 g−1).19 Hence, activation procedures are indispensable to generate highly porous ACs. ACs have been prepared from HTC-glucose by both physical activation (e.g. CO2 (ref. 10 and 21) and air22) and chemical activation (e.g. H3PO4,21 NaOH21 and KOH14,21,23–27). KOH activation is advantageous in terms of lower activation temperature, shorter activation time and higher yield.28 More importantly, this method is specifically advantageous for producing ACs for supercapacitors because of their well-defined pore size and PSD, tunable surface area and (ultra)micropore volume, and high electrical conductivity.29,30
Sevilla et al. compared the physico-chemical and electrochemical properties of ACs prepared by KOH activation of glucose and HTC-glucose.14 Using HTC-glucose, the AC yield was doubled and a significantly improved rate capability was observed for the AC-based electrodes. Recently, Sevilla et al. further optimized the KOH-ACs derived from HTC-glucose and increased the SBET from 1510 to 2950 m2 g−1, the total pore volume (Vt) from 0.68 to 1.36 cm3 g−1, and the micropore volume (VDR-N2, based on Dubinin-Radushkevich (DR) method) from 0.60 to 1.02 cm3 g−1. The optimized ACs displayed a Cm of 320 F g−1 in a two-electrode set up with a 1 M H2SO4 electrolyte.24 Notably, several other groups have prepared KOH-ACs from HTC-glucose with various degree of success when it comes to the specific capacitance (Cm). Gao et al. observed a Cm of 58 F g−1 in a 6 M KOH electrolyte for an AC with a SBET of 1197 m2 g−1, a Vt of 0.74 cm3 g−1 and a Vμp-t (micropore volume, based on t-plot method) of 0.48 cm3 g−1.25 Salinas-Torres et al. observed a Cm of 125 F g−1 in a 0.5 M H2SO4 electrolyte for an AC with a SBET of 1766 m2 g−1 and VDR-N2 of 0.58 cm3 g−1.26 Zheng et al. observed a Cm of 240 F g−1 in a 6 M KOH electrolyte for an AC with SBET of 2633 m2 g−1, a Vt of 1.86 cm3 g−1, and a Vμp-t of 0.58 cm3 g−1.27 Collectively this spread of values indicates that the Cm is strongly related to the textural properties of the ACs but also to other less obvious relations.
HTC-glucosamine has also been studied as a precursor of ACs for electrodes of supercapacitors.31 Glucosamine relates to the abundant chitin making it a viable and sustainable option. Hydrothermal carbonization of N-containing precursors is a straightforward process to enrich the carbonized products with N-containing moieties.15 This approach appears to be more straightforward than treating the precursors with ammonia or urea.32,33 N-Doped carbons also appear to have improved conductivities and enhanced capacitances due to redox reactions of electrochemically active nitrogen functional groups.32,34,35
Iron species (e.g. iron-oxide nanoparticles, Fe(NH4)2(SO4)2, FeSO4·7H2O, FeCl2·4H2O, Fe2(SO4)3) have been used as catalysts or additives in hydrothermal carbonization of biomasses.36–38 Cui et al. reported that the Fe2+ ions from Fe(NH4)2(SO4)2 and related iron-oxide nanoparticles can effectively catalyze the hydrothermal carbonization of starch and rice grains.37 Such Fe2+ ions and iron-oxide nanoparticles were ascribed structure-directing effects forming hollow microspherical and rope-like carbon-rich nanostructures, respectively. Nanoparticles of magnetic iron38 or iron oxide36 have been embedded in hydrothermal carbons and the corresponding ACs. The latter case prompts further curiosity in relation to supercapacitors as iron compounds demonstrated redox capacitance39,40 and potentially magnetic-field-enhanced capacitance.41
When designing optimal ACs for supercapacitor electrodes, it is crucial to understand the role of different types of porosity on the formation of EDL capacitance. Recent discoveries have shown that there is no simple linear correlation between the BET specific surface area (SBET) and capacitance, especially when the SBET is >1500 m2 g−1, but rather stressed the importance of optimal micropore size, pore size uniformity and pore connectivity.42 It has been proposed that ACs with very high SBET have too thin pore walls that are unable to screen completely the fields of ions in adjacent pores.43 Benchmarking studies by Gogotsi, Simon and co-workers revealed that the pores with a width of <1 nm, and close to the size of electrosorbed ions, are the most optimum for EDL formation in non-aqueous electrolyte.44–46 This new theory was further supported by Béguin's group, for both aqueous and non-aqueous electrolytes.47,48
Here, we present a systematic and comprehensive study where the textural properties (porosity, pore size, PSD etc.) of KOH-ACs derived from HTC-sugars (glucose and glucosamine) were tailored with respect to their performance as electrodes in supercapacitors. HTC-glucosamine was used to study the effect of N doping. In addition, the possible incorporation and effect of iron species from FeSO4·7H2O were investigated. Elemental composition, electrical conductivity, surface functional groups and other parameters that could be highly relevant to electrochemical performances were carefully correlated.
2. Experimental
2.1 Materials
D-(+)-Glucose (≥99.5%) [CAS number: 50-99-7], D-(+)-glucosamine hydrochloride (≥99%) [CAS number: 66-84-2], iron(II) sulfate heptahydrate (FeSO4·7H2O, ACS reagent, ≥99.0%) [CAS number: 7782-63-0], polytetrafluoroethylene (60 wt% dispersion in H2O) [CAS number: 9002-84-0], 2-propanol (≥99.8%) [CAS number: 67-63-0] were supplied by Sigma-Aldrich. Potassium hydroxide pellets (AnalaR NORMAPUR® Reag. Ph. Eur. analytical reagent) [CAS number: 1310-58-3] were obtained from VWR. Nickel meshes were purchased from Goodfellow. All chemicals were used as received.2.2 Sample preparations
Solutions of HTC-D-(+)-glucose (abbreviated as HTC-G) and HTC-D-(+)-glucosamine hydrochloride (abbreviated as HTC-GA) were prepared by dissolving the sugars in hot deionized water; amounts and additives are presented in Table 1. Typically, half of the volumes prepared were transferred and sealed in Teflon-lined stainless steel autoclaves (200 mL capacity). Then the autoclaves were heated at a temperature of 200 °C for 24 h. After the hydrothermal treatment, the autoclaves were cooled down in air by natural convection. The solid HTC-sugars were recovered by filtration, washed with deionized water and dried at 110 °C.| HTC codes | Type of sugar | Mass of sugar | FeSO4·7H2O |
|---|---|---|---|
| HTC-GFe | D-(+)-Glucose | 60 g (3.33 M) | 2.5 g (0.09 M) |
| HTC-G | D-(+)-Glucose | 60 g (3.33 M) | — |
| HTC-GAFe | D-(+)-Glucosamine HCl | 50 g (2.32 M) | 2.0 g (0.075 M) |
| HTC-GA | D-(+)-Glucosamine HCl | 50 g (2.32 M) | — |
The HTC-sugars were chemically activated by using KOH in three steps. First, the HTC-sugar was impregnated with a KOH solution (0.5 g mL−1; weight ratio of KOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HTC = 4:1) under constant stirring at room temperature overnight. Second, the mixture was dried in an oven at a temperature of 150 °C. Third, the dried and grounded mixture was thermally treated (activation) in a custom-made stainless steel reactor,36 which was placed in a vertical tube furnace. The temperature was increased at a rate of 10 °C min−1 from room temperature to the set temperature (600, 700 or 800 °C) and held there for four hours (or otherwise stated), after which the reactor was cooled down in the protective nitrogen flow (200 standard cubic centimeters per minute) used during the activation. The ACs were washed with hot deionized water until the supernatant became pH neutral and then dried at 110 °C.
:HTC = 4:1) under constant stirring at room temperature overnight. Second, the mixture was dried in an oven at a temperature of 150 °C. Third, the dried and grounded mixture was thermally treated (activation) in a custom-made stainless steel reactor,36 which was placed in a vertical tube furnace. The temperature was increased at a rate of 10 °C min−1 from room temperature to the set temperature (600, 700 or 800 °C) and held there for four hours (or otherwise stated), after which the reactor was cooled down in the protective nitrogen flow (200 standard cubic centimeters per minute) used during the activation. The ACs were washed with hot deionized water until the supernatant became pH neutral and then dried at 110 °C.
2.3 Materials characterization
The textural properties of the HTC-sugars and the corresponding ACs were studied by analyzing the volumetric N2 and CO2 adsorption–desorption isotherms recorded at −196 and 0 °C with a Micromeritics ASAP 2020 Physisorption Analyzer. Samples were degassed under dynamic vacuum conditions at temperatures of 200 (HTC-sugars) or 300 °C (ACs) for 10 h. The SBET were calculated from the N2 adsorption isotherms. Appropriate ranges of the relative pressure (p/p0) were selected to ensured positive line intersects in the multipoint BET regression analysis (C > 0), and that the composite Vads (1 − p/p0) increased with an increasing p/p0.49 The Vt was determined from the last point of adsorption isotherm at a p/p0 = 0.98.The micropore volumes and surface areas were calculated with t-plot (Vμp-t and Sμp-t) and DR (VDR-N2 and SDR-N2) methods using N2 adsorption data. The carbon black statistical thickness method (STSA) was used for the t-plot analysis. Mesopore volumes and surface areas were determined by the t-plot and Barrett–Joyner–Halenda (BJH, based on the desorption data) methods. The ultramicropore volume and surface area were calculated with the DR method using CO2 adsorption data. Throughout this manuscript, ultramicroporosity is defined as porosity with pore width less than 1 nm (predominantly 0.4–0.8 nm), accessible by CO2 molecules at 0 °C.
The average micropore width, Lo, was calculated from the characteristic energy (Eo) as determined by the DR method using the equation: Lo (nm) = 13.7/(Eo − 9.7 kJ mol−1).50 The average pore diameter Lt was estimated by 4Vt/SBET. The PSDs were calculated from N2 adsorption data using a nonlocal density functional theory (NLDFT) and carbon slit pores.
Elemental analysis of the samples was performed by the commercial company MEDAC Ltd., UK. CHN (or CHNS for some samples) composition was determined by conventional combustion analysis, while K and Fe were analyzed with a Varian Vista MPX ICP-OES system. X-ray diffraction (XRD) patterns were recorded on a Panalytical X'pert Pro alpha1 powder diffractometer in θ–2θ geometry and equipped with a Pixel detector and using a Cu-Kα1 radiation (λ: 1.5406 Å). A Perkin Elmer TGA7 was used for thermogravimetric analysis of the mass change on increasing the temperature from 20 to 900 °C at a rate of 10 °C min−1 in a flow of oxygen (a platinum cup was used). The Raman spectra were recorded by using a Horiba LabRAM HR 800 Raman spectrometer with Nd:YAG laser (532 nm/50 mW). Scanning electron microscope (SEM) images were recorded with a JEOL JSM-7000 F microscope using accelerating voltages of 5 kV. TEM were recorded with a JEOL field-emission electron microscope (JEM-2100F) operated at 200 kV, equipped with Gatan Ultrascan 1000 and Orius 200D camera. High-angle annular dark-field STEM (HAADF-STEM) images and electron energy-loss spectroscopy (EELS) spectra were acquired by JEOL ADF detector and Gatan Image Filter (GIF Tridium), respectively. The TEM specimen was prepared by dispersing a small amount of the carbon powder in ethanol. The slurry was then deposited on Cu supporting grid with holey amorphous carbon films.
For X-ray photoelectron spectroscopy (XPS), a Kratos AXIS UltraDLD spectrometer (Kratos Analytical, Manchester, UK) was used. Samples were analyzed in a monochromatic Al-based X-ray with an analysis area of ∼1 mm2 (most of the signal was from an area of 700 × 300 μm). Then the relative surface compositions (in atomic%) were obtained from quantification of detailed spectra recorded for each element. All spectra have been adjusted based on the binding energy for the carbon peak at 285.0 eV. For carbon quantification from C 1s spectra, care has been taken to subtract the overlapping K 2p peaks.
2.4 Electrochemical studies
The electrochemical behavior was studied with a Bio-Logic SP-50 potentiostat using a spring-loaded symmetrical two-electrode cell (ECC-Std electrochemical cell from EL-Cell GmbH). The electrodes were composed of 70 wt% active material (ACs), 15 wt% polytetrafluoroethylene (PTFE) and 15 wt% acetylene black. Dropwise, 2-propanol was added to the electrode composite, and the mixture was homogenized for 15 minutes with agate mortar and pestle. The paste was spread on a Ni mesh, dried, and immersed in an aqueous solution of KOH (6 mol dm−3) for at least one day to ensure a complete impregnation with the electrolyte. The loading of ACs per electrode was 5–8 mg cm−2. A laboratory qualitative filter paper (Munktell) was used as a separator.All the electrochemical studies were carried out at room temperature using a 6 M KOH electrolyte. Cyclic voltammetry (CV) and chronopotentiometry (CP) were performed in the voltage range of 0 to 1.0 V. The specific capacitance (Cm) was calculated based on two methods:
(i) CV, according to the following equation:
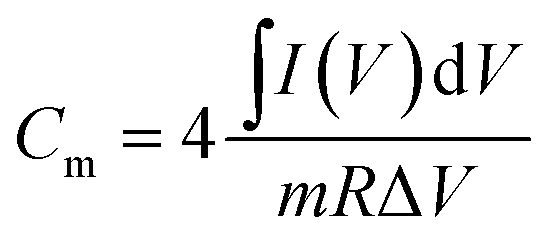
(ii) CP (discharge curve), according to the following equation:
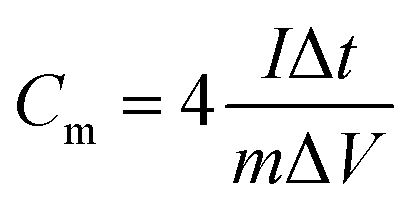
2.5 Measurement of the electrical conductivity
The electrical conductivity was measured in a purpose-built pressure cell as shown in Fig. S1 (ESI†). The cell consists of a ceramic die, and a stationary and moveable piston made of stainless steel having a diameter of 11.9 mm. The cylindrical hole of the die is 12.0 mm in diameter and the close fit creates the pressure cell. The ceramic die thus provides insulation in-between the piston and the stationary piston during measurement of the electrical conductivity.For the sample preparation, the stationary piston was placed in the die, in which the powder of AC was introduced, and the pressure cell was created by the movable piston. Pressure was applied with a Zwick universal testing machine, with a load capacity of 100 kN, and the sample was compressed stepwise with thresholds of 0.5, 2 and 7 MPa.
The DC resistance was recorded using a 4-probe micro-ohm meter Ken-MR300CA from Schuetz Messtechnik GmbH at the threshold pressure values, and once the pressure reached 7 MPa, the resistance was measured immediately and also after 1, 5 and 10 min. The conductivity was calculated based on the resistance value measured at 7 MPa after 10 min using the basic relationship σ = l/AR, where l represents the powder column height, obtained by the displacement of the piston, and A is the cross-section area of the piston.
Several reference measurements of the empty cell were made and the empty cell was found to have a resistance of 12–15 milliohm. Usually, the resistances of the compacted ACs powders were 10–50 times higher than this value. The empty-cell contribution was removed from the final data.
3. Results and discussion
3.1 Yields and compositions of HTC-G(Fe) and HTC-GA(Fe)
The yields and elemental compositions of HTC-G(Fe) and HTC-GA(Fe), with and without iron sulfate in the synthesis mixtures, are presented in Table 2. The yields of the HTCs were not influenced significantly with the addition of FeSO4·7H2O. As expected, nitrogen atoms remained in HTC-GA and HTC-GAFe (6–8 wt% N). When comparing the compositions of the HTC-G and HTC-GFe, the addition of FeSO4·7H2O did not change the CHO elemental content. Besides, only a small amount of iron was included in HTC-GFe and HTC-GAFe. Here, note that the total elemental amounts of HTC-GA(Fe) did not sum up to 100%, they reached only 85–90%, which showed that their combustion was incomplete. This difficulty for combustion was also supported by TGA, which showed that HTC-GA was more difficult to combust than HTC-G (see Fig. S2 in ESI†). After the recovery by filtration and drying, the HTC-G and HTC-GFe could easily be ground to powder, while HTC-GA and HTC-GAFe were hard and difficult to grind.| HTC | Yield (%) | Elements (wt%, dry basis) | |||||
|---|---|---|---|---|---|---|---|
| C | H | N | S | O | Fe | ||
| HTC-GFe | 47 | 65.21 | 4.54 | — | — | 29.97 | 0.18 |
| HTC-G | 44 | 66.21 | 4.27 | — | — | 29.98 | — |
| HTC-GAFe | 38 | 60.95 | 4.63 | 6.47 | 0.97 | 16.18 | 0.59 |
| HTC-GA | 40 | 56.91 | 4.97 | 7.82 | — | 14.86 | — |
3.2 Textural properties of KOH-ACs prepared from different precursors and additives
| (a) | ||||||
|---|---|---|---|---|---|---|
| AC samples | Surface area (m2 g−1) | |||||
| SBET | SDR-N2 | SDR-CO2 | Sμp-t | Sext | Smp-BJH | |
| AC-GFe-600 | 1080 | 1150 | 700 | 1025 | 55 | 50 |
| AC-GFe-700 | 1690 | 1810 | 1060 | 1635 | 55 | 50 |
| AC-GFe-750 | 2050 | 2130 | 1055 | 1910 | 140 | 130 |
| AC-GFe-800-2h | 2410 | 2500 | 1130 | 2190 | 220 | 165 |
| AC-GFe-800 | 2260 | 2250 | 765 | 1940 | 320 | 230 |
| AC-G-600 | 880 | 935 | 840 | 845 | 35 | 30 |
| AC-G-700 | 1300 | 1380 | 1160 | 1250 | 50 | 40 |
| AC-G-800 | 1550 | 1610 | 825 | 1415 | 135 | 125 |
| AC-GAFe-600 | 1715 | 1820 | 1070 | 1620 | 95 | 80 |
| AC-GAFe-700 | 2320 | 2300 | 770 | 2110 | 210 | 170 |
| AC-GAFe-800 | 2355 | 2230 | 670 | 1965 | 390 | 350 |
| AC-GA-600 | 1930 | 1990 | 960 | 1790 | 140 | 130 |
| AC-GA-700 | 3000 | 2920 | 795 | 2540 | 460 | 375 |
| AC-GA-800 | 3080 | 2990 | 640 | 1340 | 1740 | 1220 |
| Norit SX ultra | 985 | 1020 | 590 | 740 | 245 | 265 |
| (b) | |||||||
|---|---|---|---|---|---|---|---|
| ACs | Pore volume (cm3 g−1) | Ave. pore size (nm) | |||||
| Vt | Vμp-t | Vmp-BJH | VDR-N2 | VDR-CO2 | Lt | Lo | |
| AC-GFe-600 | 0.455 | 0.374 | 0.062 | 0.408 | 0.282 | 1.69 | 0.69 |
| AC-GFe-700 | 0.679 | 0.596 | 0.055 | 0.643 | 0.426 | 1.60 | 0.77 |
| AC-GFe-750 | 0.888 | 0.681 | 0.127 | 0.757 | 0.422 | 1.73 | 1.06 |
| AC-GFe-800-2h | 1.043 | 0.795 | 0.153 | 0.889 | 0.454 | 1.73 | 1.09 |
| AC-GFe-800 | 1.026 | 0.668 | 0.196 | 0.799 | 0.306 | 1.82 | 1.23 |
| AC-G-600 | 0.356 | 0.309 | 0.030 | 0.332 | 0.337 | 1.62 | 0.58 |
| AC-G-700 | 0.520 | 0.455 | 0.043 | 0.489 | 0.466 | 1.61 | 0.64 |
| AC-G-800 | 0.685 | 0.500 | 0.115 | 0.573 | 0.331 | 1.77 | 0.98 |
| AC-GAFe-600 | 0.713 | 0.586 | 0.065 | 0.647 | 0.427 | 1.66 | 0.97 |
| AC-GAFe-700 | 1.003 | 0.721 | 0.138 | 0.816 | 0.307 | 1.73 | 1.42 |
| AC-GAFe-800 | 1.089 | 0.588 | 0.259 | 0.793 | 0.267 | 1.85 | 1.36 |
| AC-GA-600 | 0.830 | 0.627 | 0.101 | 0.707 | 0.386 | 1.72 | 1.09 |
| AC-GA-700 | 1.368 | 0.780 | 0.282 | 1.037 | 0.319 | 1.82 | 1.53 |
| AC-GA-800 | 1.704 | 0.192 | 0.920 | 1.063 | 0.257 | 2.21 | 2.06 |
| Norit SX ultra | 0.743 | 0.271 | 0.461 | 0.364 | 0.238 | 3.02 | 1.07 |
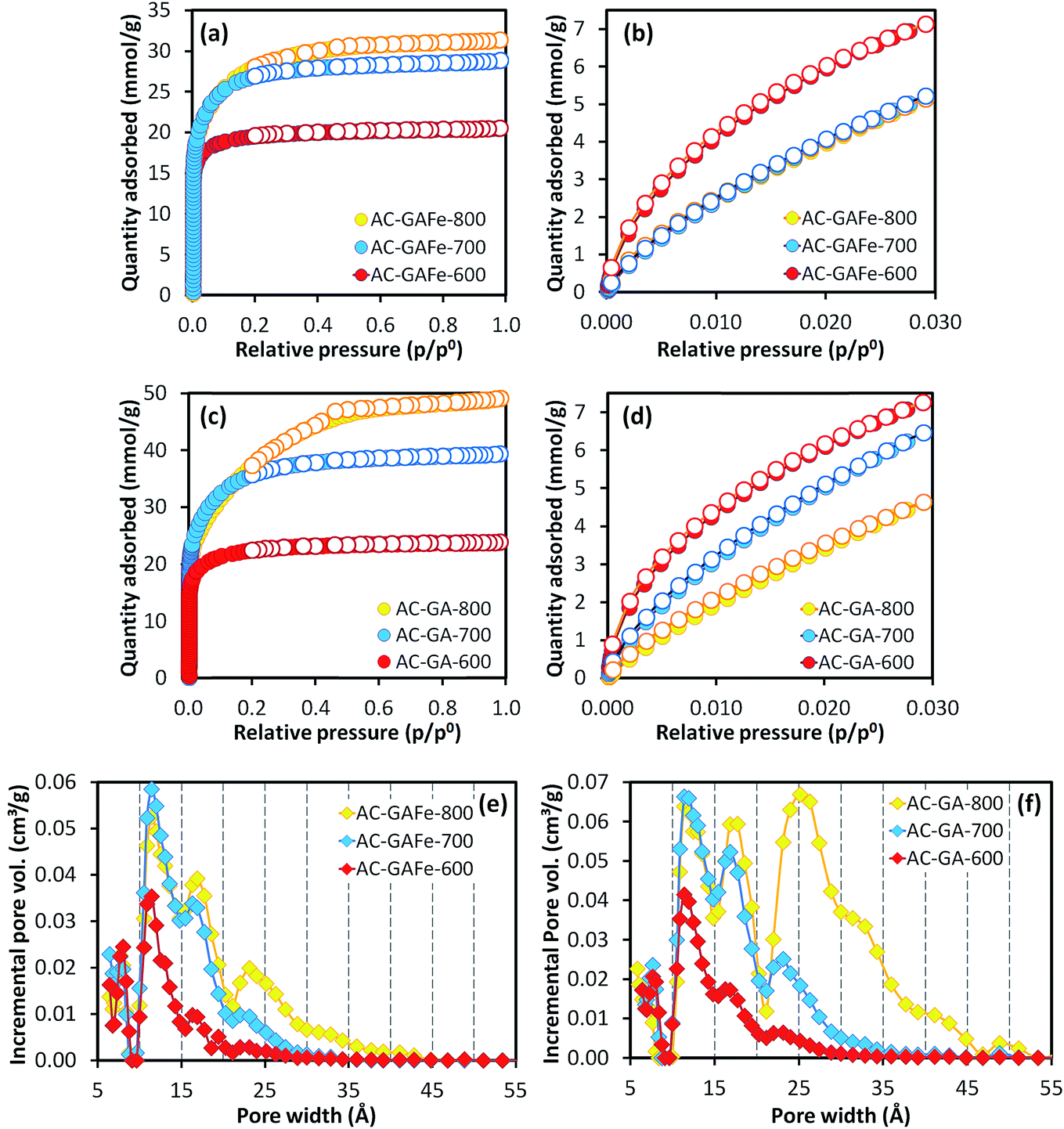
A high activation temperature has been shown to promote the formation of mesopores for KOH-ACs,47,52 which we also observed. The AC-GFe-600 and AC-GFe-700 had N2 isotherms (Fig. 1a and c) typical for micropores (type 1),53,54 and narrow PSDs with peaks at 8 and 11 Å (Fig. 1e). The AC-GFe-800 and AC-G-800, on the other hand, displayed additional mesopores (compare PSDs in Fig. 1e and f). Distinctively, AC-GFe-800 and AC-G-800 had additional pores in the range of 15 to 30 Å. The AC-GFe-750 exhibited a transitional PSD type between AC-GFe-700 and AC-GFe-800.
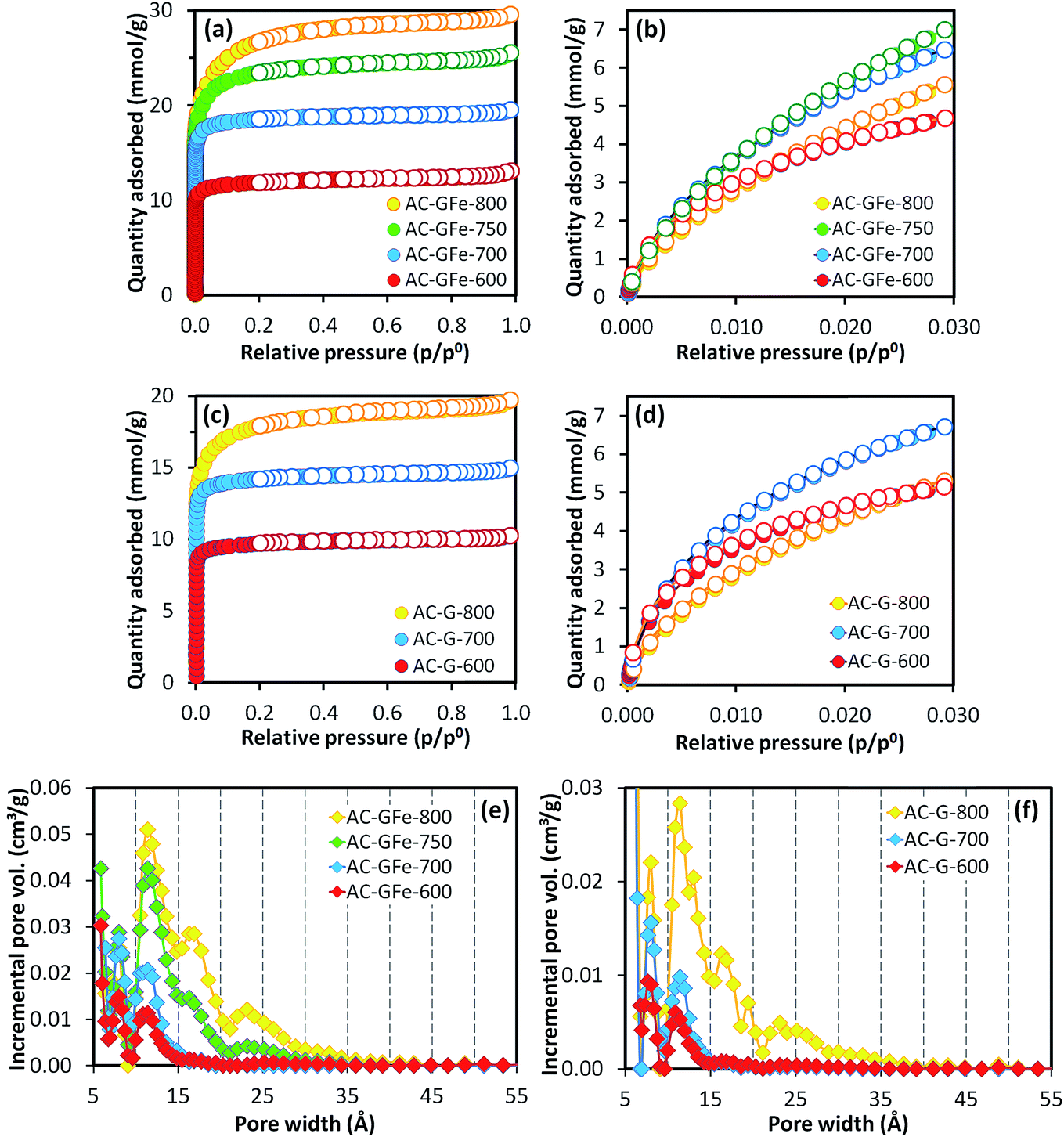 | ||
| Fig. 1 (a and c) N2 adsorption–desorption isotherms at −196 °C and (b and d) CO2 adsorption–desorption isotherms at 0 °C for AC-GFe and AC-G series. The absolute pressure for CO2 adsorption ranged from 0.3 to 102 kPa. Filled and empty symbols represent adsorption and desorption, respectively. Pore size distributions of AC-GFe (e) and AC-G (f) computed based on N2 adsorption isotherm at −196 °C on carbon slit pores by NLDFT. All ACs were activated for 4 h. Activated carbons (AC-X-Y) from HTC-glucose with and without iron (X: G or GFe) prepared at temperatures of Y: 600–800 °C. | ||
The transition between PSD types can be rationalized from the reactions of KOH with the carbon precursor mixture at elevated temperature (see ref. 28 for details on the mechanism of KOH activation). In brief, the massive release of various gaseous species above 700 °C, etches the carbon framework together with various potassium compounds. It also potentially contributes to intercalation of metallic K into the carbon lattices.55 At these high temperatures, ACs with comparably large pores are formed, as is shown by our experiments.
The ultramicroporosity (as probed by CO2 sorption) showed a maximum with respect to the activation temperature for both the AC-GFe and AC-G series. The highest ultramicropore area (SDR-CO2) was reached at the intermediate activation temperature of 700 °C. Both AC-GFe-700 and AC-G-700 had a high SDR-CO2 of 1000–1200 m2 g−1 and a high ultramicropore volume (VDR-CO2) of 0.42–0.47 cm3 g−1, while the ACs formed at 800 and at 600 °C had significantly lower SDR-CO2 and VDR-CO2 (Table 3). In other words, a high activation temperature of 800 °C mainly contributed to a high pore volume in the range of wider micropore (>1 nm) and small mesopores (2–3 nm).
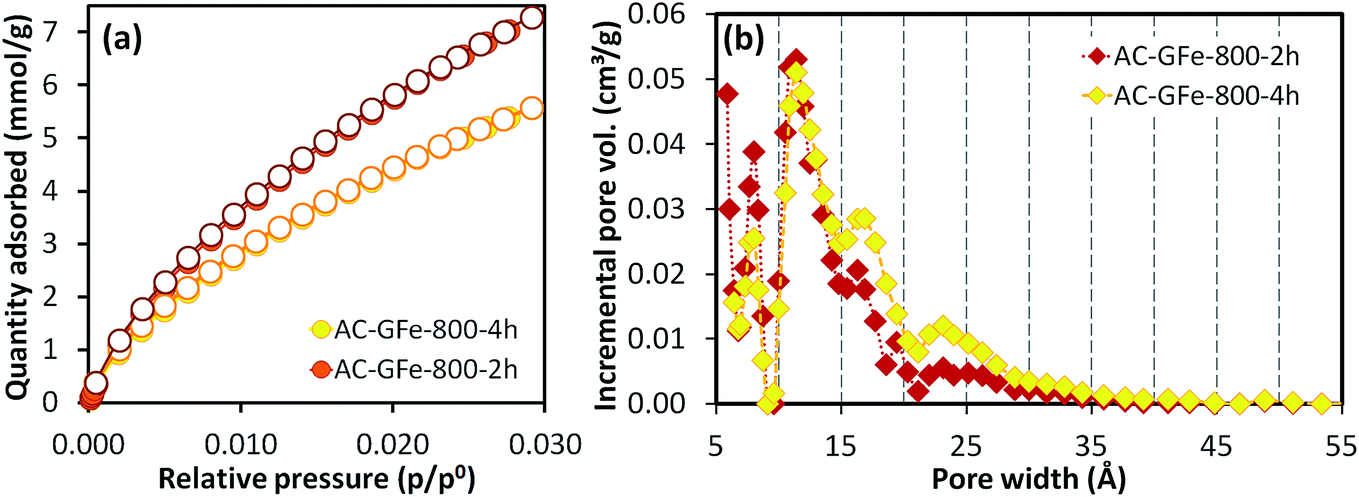 | ||
| Fig. 2 (a) CO2 adsorption–desorption isotherms at 0 °C for AC-GFe-800-2h and AC-GFe-800-4h, the absolute pressure for CO2 adsorption ranged from 0.3 to 102 kPa; (b) pore size distributions show that AC-GFe-800-2h possesses higher ultramicropore (<1 nm) volume. Activated carbons (AC-X-Y) from hydrothermally carbonized glucose with iron (X: GFe) prepared at a temperature of Y: 800 °C. | ||
The N2 isotherms of AC-GFe-800-2h and AC-GFe-800-4h were, on the other hand, almost identical as shown in the Fig. S3,† and the SBET and Vt of AC-GFe-800-2h (2414 m2 g−1 and 1.043 cm3 g−1) were only slightly higher than those of AC-GFe-800-4h (2260 m2 g−1 and 1.026 cm3 g−1). The AC-GFe-800-2h showed a higher pore volume at the pore width of <10 Å than AC-GFe-800-4h. Meanwhile, AC-GFe-800-4h showed a higher pore volume at the pore width of 15–35 Å than AC-GFe-800-2h (Fig. 2b). The higher surface area and pore volume of AC-GFe-800-4h in the range of wider micropore and mesopore were confirmed by t-plot and BJH analysis (Table 3).
:HTC ratio, activation temperature and duration, our equivalent ACs showed SBET values of 1716 (AC-GAFe-600) and 1930 (AC-GA-600) m2 g−1, presenting an impressive three-fold increment. This marked improvement is probably due to a better mixing of KOH and HTC-GA (or HTC-GAFe) by the wet impregnation method as compared to the physical mixing used in ref. 31.
 | ||
| Fig. 3 (a and c) N2 adsorption–desorption isotherms at −196 °C and (b and d) CO2 adsorption–desorption isotherms at 0 °C for AC-GAFe and AC-GA series. The absolute pressure for CO2 adsorption ranged from 0.3 to 102 kPa. Filled and empty symbols represent adsorption and desorption, respectively. Pore size distributions of AC-GAFe (e) and AC-GA (f) series computed based on N2 adsorption data recorded at a temperature of −196 °C using a nonlocal density functional theory based on carbon slit pores. All ACs were activated for 4 h. Activated carbons (AC-X-Y) from hydrothermally carbonized glucosamine with and without iron (X: GA or GAFe) prepared at temperatures of Y: 600–800 °C. | ||
Interestingly, the AC-GA-600 and AC-GAFe-600 (Fig. 3e and f) displayed PSDs similar to AC-G-800 and AC-GFe-800 rather than the 600 series. Tentatively, we ascribe this change to the chemical difference of HTC-G and HTC-GA. As mentioned earlier, there are additional nitrogen-containing chemical groups (pyrrole-like molecules, amide bonds and aromatic amines) in HTC-GA.15 Different amounts and types of gaseous species would be generated during the thermal treatment for HTC-G and HTC-GA, which appear to have led to a generation of porosity at a lower temperature for HTC-GA. Further investigations are required to fully understand the complicated reaction mechanisms occurring during the activation of HTC-GA and HTC-GAFe; however, these were not the main focus of this study.
Irrespective of the detailed mechanisms of activation, the AC-GA and AC-GAFe series demonstrated higher specific surface areas and wider PSDs than the AC-G and AC-GFe series. From Table 3b, it is clear that the AC-GA and AC-GAFe series had larger average pore diameters (Lt) and average micropore widths (Lo) than the AC-G and AC-GFe series. As for the SBET, the ultramicroporosity of the AC-GA and AC-GAFe series peaked at a lower activation temperature than for the AC-G and AC-GFe series. For the AC-GA and AC-GAFe series, the highest SDR-CO2 and VDR-CO2 occurred at 600 °C.
3.3 Elemental composition of activated carbons
In the XPS analysis of selected ACs, wide (survey) spectra as shown in Fig. S4 and S5† were recorded to detect the elements of the surface layer of selected ACs. The shape of all high-resolved carbon spectra resembles that of carbon black. Detailed assignment of the bands was not possible due to different oxidation states indicated by the very long slope on the high binding energy side of the peaks in the spectra (Fig. S6 and S7†).The carbon content and associated O/C atomic ratio decreased with an increased activation temperature for all the series of ACs. As shown in Table 4, the carbon content of AC-GFe-800-2h and AC-GA-800 was 90.6 and 91.8 at%, compared to 85.7 and 83.4 at% for AC-GFe-700 and AC-GA-700. Meanwhile, the oxygen content of AC-GFe-800-2h and AC-GA-800 was 7.8 and 6.1 at%, compared to 11.0 and 11.8 at% for AC-GFe-700 and AC-GA-700. The bulk elemental compositions of carbon and oxygen for the ACs are presented in Fig. S8,† which show a trend of increased carbon and decreased oxygen content on increased activation temperature. The AC-GA and AC-GAFe series contained 0.8–1.5 at% of N based on XPS analysis (Table 4). At a higher temperature, more nitrogen was released.
| Sample | Atomic% | |||||||
|---|---|---|---|---|---|---|---|---|
| C | O | N | Fe | K | S | Si | Ni | |
| AC-G-700 | 89.7 | 9.0 | — | — | 1.2 | — | 0.1 | — |
| AC-GFe-700 | 85.7 | 11.0 | — | 1.4 | 1.6 | 0.1 | (0.1) | (0.1) |
| AC-GFe-800-2h | 90.6 | 7.8 | 0.4 | 0.2 | 0.8 | 0.1 | (0.1) | — |
| AC-GA-700 | 83.4 | 11.8 | 1.5 | 0.3 | 3.0 | <0.05 | — | — |
| AC-GA-800 | 91.8 | 6.1 | 0.8 | 0.3 | 0.9 | — | — | — |
| AC-GAFe-700 | 71.4 | 16.8 | 1.1 | 9.2 | 0.8 | 0.1 | 0.4 | 0.2 |
For the AC-GFe series (Fig. S9c†) and the AC-GAFe-600 (Fig. S9e†), the XRD patterns corresponded to Fe3O4 and Fe. However, only metallic Fe was observed for the AC-GAFe-700 and AC-GAFe-800. Unexpectedly, contamination by metallic Fe was detected in all other ACs. The origin of the Fe contaminants was the corrosion from the stainless steel mesh used in our reactor. To avoid Fe contaminants in future studies, a base-resistant nickel mesh is suggested. The metallic impurities can also be minimized by acid washing (e.g. HCl) after KOH activation.
3.4 Electrochemical studies
The electrochemical performance of the ACs was studied in a symmetrical two-electrode cell using a 6 M KOH electrolyte. The two-electrode cell mimicked the physical configuration, internal voltages and charge transfer that occur in real supercapacitor devices.56 Consequently, it gives the best and most reliable indication of the performance of the ACs as electrode materials. A commonly available commercial AC, Norit SX Ultra, was used as a reference material and served as an internal validation of the electrochemical measurements (see Table 3 for textural properties).Notably, all of the HTC-sugar-based ACs had higher specific capacitances (Cm) than Norit SX Ultra (Fig. 4 and Table 5) as determined by cyclic voltammetry (CV) and chronopotentiometry (galvanostatic charge–discharge studies) (Fig. S10† and Table 5). The maximum Cm values were determined at a low scan rate (2 mV s−1) (Table 5), and the Cm decreased on increasing scan rates (up to 200 mV s−1). At 2 mV s−1, peak currents associated with oxidation at 1 V can be observed for all samples (Fig. 4 and 6). To evaluate the effect of the oxidation on the calculation of Cm, we also calculate the Cm using the voltage range from 0 to 0.90 V and 0 to 0.80 V. The values (Table S1†) show that the extent of oxidation does not significantly affect the Cm values; albeit the Cm values of samples AC-GA-600 and AC-GAFe-600 increased more significantly than other samples because of their lower capacitance in 0.9 to 1.0 V (to be discussed in the latter section). Furthermore, it was noted that the oxidation subsided on increasing scan rates. For instance, CV curves of sample AC-G-700 at different rates are presented in Fig. S11.†
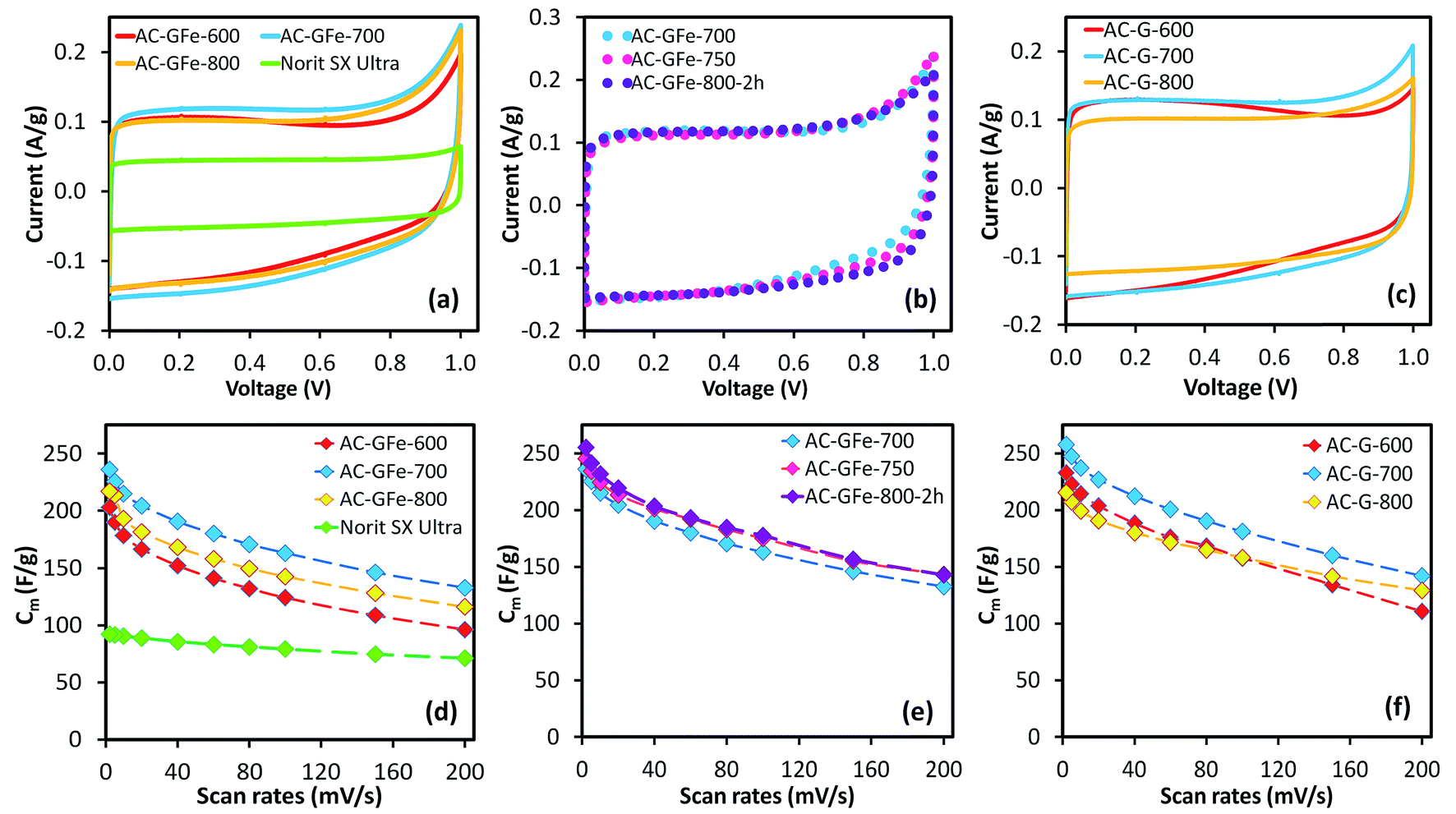 | ||
| Fig. 4 (a–c) Cyclic voltammetry (CV) curves of AC-GFe and AC-G series at 2 mV s−1, and (d–f) specific capacitances (Cm) calculated from CV scanned from 2 to 200 mV s−1. Activated carbon (AC-X-Y) from hydrothermally carbonized glucose with and without iron (X: G, GA, GFe, or GAFe) prepared at temperatures of Y: 600–800 °C for 4 h (except 2 h for AC-GFe-800-2h). | ||
| ACs | Specific capacitance Cm (F g−1) | Rate performance (%) | ||
|---|---|---|---|---|
| CV, 2 mV s−1 | CV, 200 mV s−1 | CP, 0.2 A g−1 | ||
| AC-GFe-600 | 203 | 96 | 193 | 47% |
| AC-GFe-700 | 241 | 133 | 232 | 56% |
| AC-GFe-750 | 246 | 143 | 234 | 58% |
| AC-GFe-800-2h | 255 | 143 | 240 | 56% |
| AC-GFe-800 | 217 | 116 | 208 | 54% |
| AC-G-600 | 233 | 111 | 227 | 48% |
| AC-G-700 | 258 | 142 | 247 | 55% |
| AC-G-800 | 215 | 129 | 204 | 60% |
| AC-GAFe-600 | 243 | 103 | 233 | 42% |
| AC-GAFe-700 | 251 | 112 | 242 | 45% |
| AC-GAFe-800 | 230 | 139 | 226 | 61% |
| AC-GA-600 | 215 | 120 | 203 | 56% |
| AC-GA-700 | 263 | 154 | 250 | 58% |
| AC-GA-800 | 226 | 131 | 214 | 58% |
| Norit SX ultra | 92 | 71 | 84 | 77% |
Retaining the highest possible Cm at high scan rates (i.e. rate performance) is critical in developing supercapacitor electrodes57–59 as it directly affects the power output. The best performers in this study retained 140–155 F g−1 at scan rates of 200 mV s−1 as shown in Table 5. At a low scan rate, the ions can effectively penetrate the pores and increase the number of charges in the EDL leading to a high Cm. For more rapid scan rates, the diffusion limitation of the electrolyte in the pores reduces the Cm.60 Generally, the rate performance improved when activation temperature increased from 600 to 800 °C, where an increased activation temperature was shown to widen the PSD (Fig. 1e and f and 3e and f). Beside diffusion limitation, ohmic resistances of the electrolyte or the ACs can reduce the Cm.30,60 Different electrical conductivities of the ACs (Fig. S13†) did not significantly impact the rate performance. This can be related to the electrode composition (70% AC: 15% carbon black: 15% PTFE). The high content of binder and conductive additive equalized the conductivities of the electrodes, thus limited the influence from the differential conductivity of the ACs.
The Cm of the AC-G and AC-GFe series were in the range of 200–260 F g−1, and the highest Cm was recorded for those prepared at 700 °C: 241 F g−1 for AC-GFe-700 and 258 F g−1 for AC-G-700 (Fig. 4a and c). The values for Cm of the AC-G and AC-GFe series correlated with the ultramicropore parameters (SDR-CO2 and VDR-CO2) but not with the SBET (Fig. 5). These tendencies are consistent with the findings of Vix-Guterl et al., who showed that the Cm of templated porous carbons in both aqueous and organic electrolytes were proportional to the CO2-based ultramicropore volume.48 Besides, Raymundo-Piñero et al. showed an increasing efficiency of the pores in ACs for forming the EDLs in aqueous electrolytes of 6 M KOH or 1 M H2SO4 when pore size decreases from 1.4 to 0.7 nm.47 In other words, the optimal pore size for ACs used with aqueous electrolytes is around 0.7 nm. The reason for the enhancement of capacitance in the region of ultramicroporosity was initially explained by Chmiola et al. The distortion of the ion solvation shell and partial removal of the ion solvation in the ultramicropores would reduce the distance between ion and carbon surface (also known as charge separation distance) and thus improve the electric double layer capacitance. As a result, the specific capacitance is correlated closely to ultramicroporosity probed by CO2 adsorption.44,61
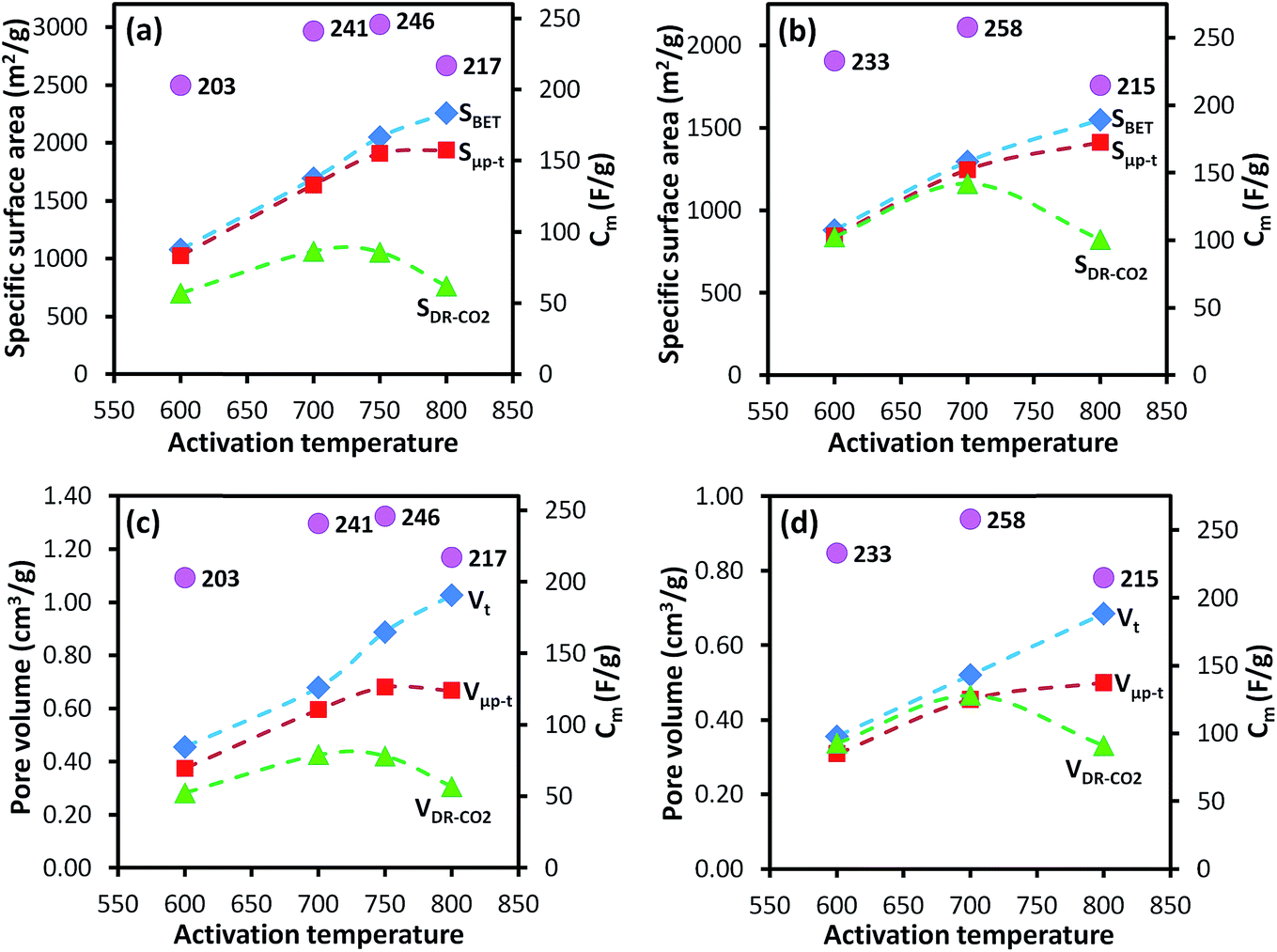 | ||
| Fig. 5 Relationship between activation temperature vs. specific surface areas and pore volumes (dashed lines) calculated by various methods, and Cm (purple dots) for AC-GFe (a and c) and AC-G (b and d) series. Activated carbon (AC-X) from hydrothermally carbonized glucose with and without iron (X: G or GFe). | ||
These findings support previous studies which have shown that the Cm of ACs do not correlate linearly with the SBET, especially when the SBET is higher than 1200–1500 m2 g−1.1,62,63 On the contrary, a high Cm of ∼260 F g−1 is achievable at a moderately high SBET of ∼1300 m2 g−1 (AC-G-700), equivalent to Cm/SBET of 20 μF cm−2, which is among the highest value reported for undoped porous carbons. From an industrial perspective, a SBET between 1000–1500 m2 g−1 of ACs appears to be desirable.64
While the CV curve of the Norit SX Ultra exhibited an ideal rectangular shape, which is generally noticed for EDL capacitors with low internal resistance, the HTC-sugar derived ACs displayed slight deviations from the rectangular shape (Fig. 4). This deviation could be attributed to the oxygen rich nature of the ACs. Furthermore, the surface oxygen functional groups may have contributed to the increased responses of the current density when the voltage approached 1 V, which correlated with the hydrolysis of water.
The shape of the CV curves for AC-G-600 and AC-GFe-600 presented in Fig. 4a(i) and c(i) deviated significantly from the ideal rectangular shape. During the voltage cycling, a higher current was observed at low voltage (<0.4 V) but a comparably lower current was noticed at higher voltage (>0.4 V). This behavior is unconventional for EDL capacitance. A possible explanation could be an “ion-sieving” effect, which could occur when the sizes of electrolyte ions are larger than the pore openings.65–67 However, we observed similar CV curves for AC-GA-600 and AC-GAFe-600 (Fig. 6a and b(i)), which have comparably larger pores (see Section 3.2) and hence it rules out the “ion sieving” effect in this case. Instead, the particular higher current responses at low voltage range could be assigned to pseudocapacitance derived from oxygen functional groups at the surface of the ACs prepared at 600 °C. These ACs have a considerably higher oxygen content (and lower carbon content) than the ACs prepared at higher temperatures (see Section 3.3). Such features in the CV curve were observed for graphene oxide (carbon with rich oxygen functional groups) in a comparative study of graphene (relatively “pure” carbon) and graphene oxide as electrode materials for supercapacitors.68 A higher activation temperature ≥700 °C reduces the density of surface functionalities on the ACs and these ACs resemble ore closely to the rectangular-shaped CV of EDL capacitors.30
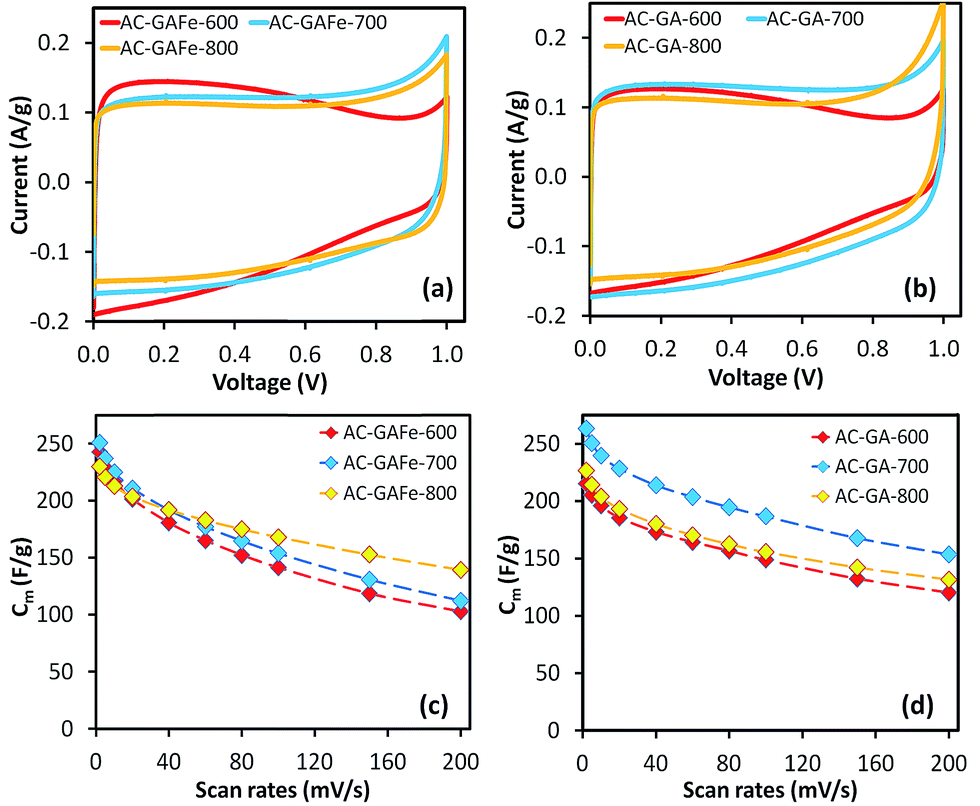 | ||
| Fig. 6 (a and b) Cyclic voltammetry (CV) curves of AC-GA and AC-GAFe series at 2 mV s−1, and (c and d) specific capacitances (Cm) calculated from CV scanned from 2 to 200 mV s−1. Activated carbon (AC-X-Y) from hydrothermally carbonized glucosamine with and without iron (X: GA or GAFe) prepared at temperatures of Y: 600–800 °C. | ||
The cycle life of an electrochemical cell based on electrode AC-G-700 was evaluated by repeating CV scans at 100 mV s−1 over 0 to 1 V for 10000 cycles (Fig. S14†). The calculated Cm derived from the CV curves at different cycles were normalized to the value of an earlier cycle (cycle 5) in Fig. S14.† The capacitance of the electrochemical cell increased slightly (3%) after 1000 cycles. After that, the value remained stable up to 10000 cycles.
For the AC-GA and AC-GA-Fe series prepared from HTC-GA, the Cm were 215 to 263 F g−1 (cf. Fig. 6) but no clear-cut correlations of the Cm with the SDR-CO2 or VDR-CO2 were observed (Fig. 7). Nonetheless, the optimum activation temperature with respect to the Cm (AC-GA and AC-GAFe series) was 700 °C. This optimum temperature can be rationalized with respect to several possible reasons e.g. the surface area, carbon content and electrical conductivity. When comparing the CV curves of AC-GAFe-600 and AC-GAFe-700 in Fig. 6a, the additional oxygen functional groups of AC-GAFe-600 appeared to contribute with additional pseudocapacitance in the low voltage range. However, that was not sufficient to compensate for the loss of EDL capacitance at the high voltage range. The lower carbon content and extremely low electrical conductivity (Fig. S13†) of AC-GA-600 and AC-GAFe-600 restricted their EDL formation although they possessed values of Sμp-CO2 comparable to the best ACs in the AC-G and AC-GFe series. The AC-GA-700 and AC-GAFe-700 performed generally better than AC-GA-800 and AC-GAFe-800 albeit their similar values of SBET.
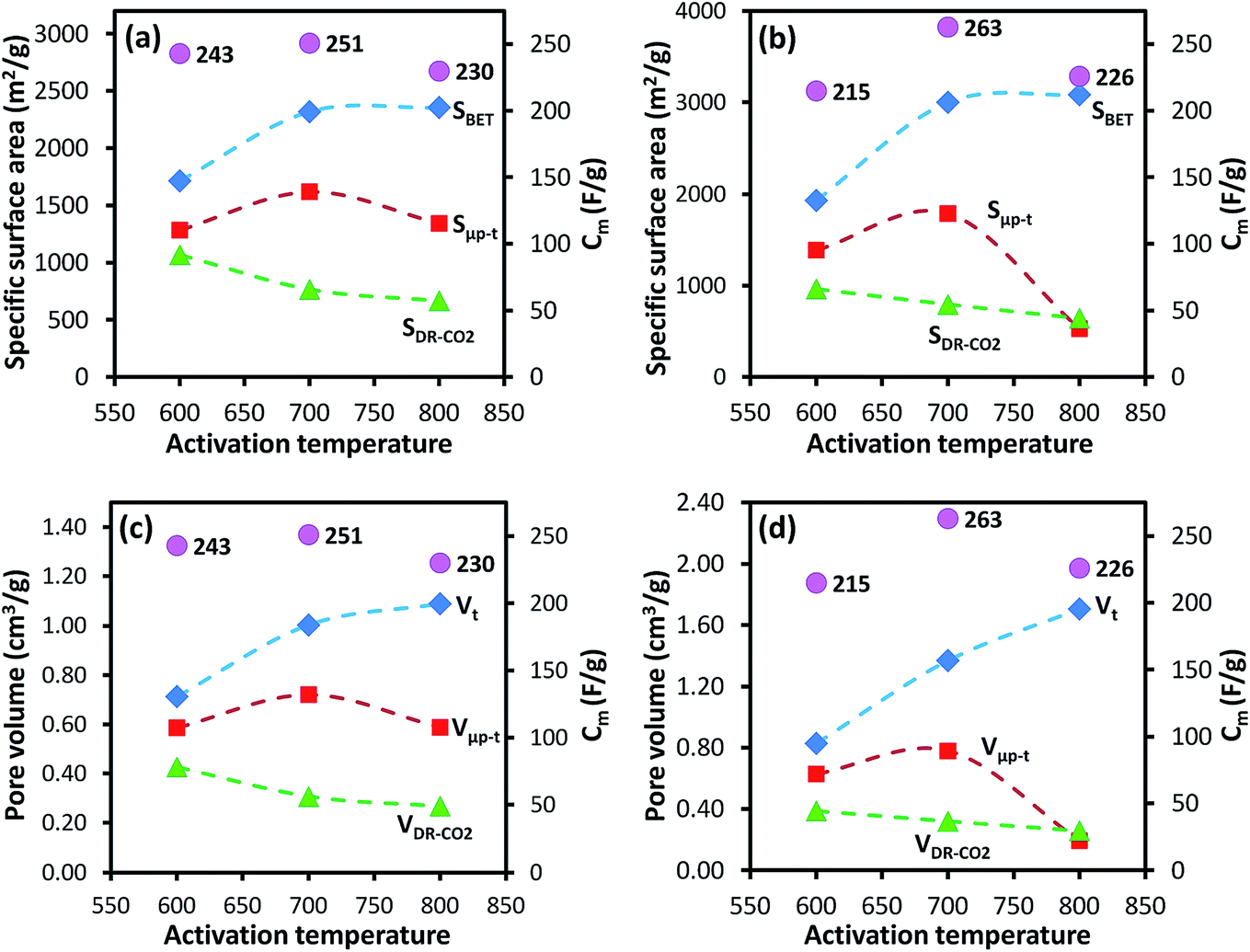 | ||
| Fig. 7 Relationship between activation temperature vs. specific surface areas and pore volumes (dashed lines) calculated by various methods, and Cm (purple dots) for AC-GAFe (a and c) and AC-GA (b and d) series. Activated carbon (AC-X) from hydrothermally carbonized glucosamine with and without iron (X: GA or GAFe). | ||
The electrical conductivity of the AC-GA and AC-GAFe series were unexpectedly much lower than their AC-G and AC-GFe counterparts (Fig. S13†). As mentioned in the introduction, N-doping is widely reported as a strategy to enhance the electrical conductivity of carbon materials. The relatively low electrical conductivity of the AC-GA and AC-GAFe series can be correlated to the destruction of the structural order of the carbons (with corresponding very high SBET up to 3000 m2 g−1 and higher mesoporosity). In a similar manner, the N-doped ACs with mesoporosity prepared by Sevilla et al. possessed lower electrical conductivity (120–130 S m−1) as compared with their undoped analogues (450–460 S m−1).24
Further optimization of AC-GA and AC-GAFe would be required (e.g. by lowering the KOH:HTC ratio) to more conclusively correlate the effect of porosity in the N-doped ACs to capacitances. The direction of the optimization would include preparing N-doped ACs with a narrow PSD but with different extent of ultramicroporosity, and ACs with similar surface area and pore volumes but with different average pore sizes and average micropore widths.
4. Conclusions
Textural properties (specific surface areas for different type of porosity, porosity from ultramicropores to supermicropores to mesopores) of ACs derived from HTC-G and HTC-GA could be finely tuned by changing the precursor mixtures and KOH-activation conditions (e.g. temperature, duration). All ACs were rich in oxygen functional groups, and the ACs derived from HTC-GA were enriched in nitrogen as well. This control opens up for further optimization of ACs derived from these and related precursors. The PSDs of AC-G(Fe) series were controlled by the activation temperature. At temperatures ≤700 °C, the ACs had narrow PSD with predominantly ultramicropores (Sμp-CO2/SBET ≈ 62–65%) and at >700 °C, larger pores (Sμp-CO2/SBET ≈ 30–52%) were formed due to the decomposition of K2CO3. The Cm of the AC-GFe and AC-G series in an aqueous electrolyte, 6 M KOH, correlated strongly with the ultramicroporosities that were probed by CO2 physisorption. Albeit the surface specific capacitances in the highly porous ACs were very high (20 μF cm−2), we foresee a strong need to further increase the rate capabilities of this kind of AC-based supercapacitor electrodes. By even higher activation temperatures the carbon content will be increased (lower O/C), and thus the electrical conductivity would be increased. It could certainly be relevant to further study the possibilities to increase the rate capabilities, while maintaining a high ultramicroporosity that appears to be a key to the function of carbon based EDL capacitors in aqueous electrolytes.Acknowledgements
This project (No. 36912-2) was supported by the Swedish Energy Agency (Batterifondprogrammet). N. Hedin wishes to thank the award of postdoctoral fellowship (CTS 15:203) for K. K. Lee from the Carl Trygger Foundation for Scientific Research.References
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS PubMed.
- T. D. Atmaja and Amin, Energy Procedia, 2015, 68, 429–437 CrossRef CAS.
- J. W. Long, Electrochem. Soc. Interface, 2008, 17, 33 CAS.
- J. R. Miller and A. F. Burke, Electrochem. Soc. Interface, 2008, 17, 53–57 CAS.
- A. Burke, Int. J. Energy Res., 2010, 34, 133–151 CrossRef CAS.
- A. Burke, Electrochim. Acta, 2007, 53, 1083–1091 CrossRef CAS.
- S. Dietz and D. Recla, Proceedings of the 14th International Seminar on Double Layer Capacitors and Hybrid Energy Storage Devices Florida, 2004 Search PubMed.
- S. Dietz and D. Recla, US7541312 B2, 2009.
- S.-E. Chun, Y. N. Picard and J. F. Whitacre, J. Electrochem. Soc., 2011, 158, A83–A92 CrossRef CAS.
- T. Thomberg, T. Tooming, T. Romann, R. Palm, A. Janes and E. Lust, J. Electrochem. Soc., 2013, 160, A1834–A1841 CrossRef CAS.
- M.-M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 Search PubMed.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- M. M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- M. Sevilla, L. Yu, C. O. Ania and M.-M. Titirici, ChemElectroChem, 2014, 1, 2138–2145 CrossRef CAS.
- L. Zhao, N. Baccile, S. Gross, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Carbon, 2010, 48, 3778–3787 CrossRef CAS.
- X. Fan, C. Yu, Z. Ling, J. Yang and J. Qiu, ACS Appl. Mater. Interfaces, 2013, 5, 2104–2110 CAS.
- Z. Jinli, W. Jiao, L. Yuanyuan, N. Ning, G. Junjie, Y. Feng and L. Wei, J. Mater. Chem. A, 2015, 3, 2043–2049 Search PubMed.
- J. Wu, C. Jin, Z. Yang, J. Tian and R. Yang, Carbon, 2015, 82, 562–571 CrossRef CAS.
- Q. Wang, H. Li, L. Chen and X. Huang, Carbon, 2001, 39, 2211–2214 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- A. J. Romero-Anaya, M. Ouzzine, M. A. Lillo-Ródenas and A. Linares-Solano, Carbon, 2014, 68, 296–307 CrossRef CAS.
- Y. Gong, H. Wang, Z. Wei, L. Xie and Y. Wang, ACS Sustainable Chem. Eng., 2014, 2, 2435–2441 CrossRef CAS.
- C. Falco, J. P. Marco-Lozar, D. Salinas-Torres, E. Morallón, D. Cazorla-Amorós, M. M. Titirici and D. Lozano-Castelló, Carbon, 2013, 62, 346–355 CrossRef CAS.
- A. B. Fuertes and M. Sevilla, ChemSusChem, 2015, 8, 1049–1057 CrossRef CAS PubMed.
- F. Gao, G. Shao, J. Qu, S. Lv, Y. Li and M. Wu, Electrochim. Acta, 2015, 155, 201–208 CrossRef CAS.
- D. Salinas-Torres, D. Lozano-Castello, M. M. Titirici, L. Zhao, L. Yu, E. Morallon and D. Cazorla-Amoros, J. Mater. Chem. A, 2015, 3, 15558–15567 CAS.
- X. Zheng, W. Lv, Y. Tao, J. Shao, C. Zhang, D. Liu, J. Luo, D.-W. Wang and Q.-H. Yang, Chem. Mater., 2014, 26, 6896–6903 CrossRef CAS.
- J. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC.
- M. Olivares-Marín, J. A. Fernández, M. J. Lázaro, C. Fernández-González, A. Macías-García, V. Gómez-Serrano, F. Stoeckli and T. A. Centeno, Mater. Chem. Phys., 2009, 114, 323–327 CrossRef.
- J. Sánchez-González, F. Stoeckli and T. A. Centeno, J. Electroanal. Chem., 2011, 657, 176–180 CrossRef.
- L. Zhao, L. Z. Fan, M. Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS PubMed.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- P. Burg, P. Fydrych, D. Cagniant, G. Nanse, J. Bimer and A. Jankowska, Carbon, 2002, 40, 1521–1531 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- G. Wang, J. Zhang, S. Kuang, J. Zhou, W. Xing and S. Zhuo, Electrochim. Acta, 2015, 153, 273–279 CrossRef CAS.
- W. Hao, E. Björkman, Y. Yun, M. Lilliestråle and N. Hedin, ChemSusChem, 2014, 7, 875–882 CrossRef CAS PubMed.
- X. Cui, M. Antonietti and S. H. Yu, Small, 2006, 2, 756–759 CrossRef CAS PubMed.
- Q. Yan, J. Street and F. Yu, Biomass Bioenergy, 2015, 83, 85–95 CrossRef CAS.
- K. K. Lee, S. Deng, H. M. Fan, S. Mhaisalkar, H. R. Tan, E. S. Tok, K. P. Loh, W. S. Chin and C. H. Sow, Nanoscale, 2012, 4, 2958–2961 RSC.
- K. K. Lee, R. W. Y. Ng, K. K. She, W. S. Chin and C. H. Sow, Mater. Lett., 2014, 118, 150–153 CrossRef CAS.
- J. Zhu, M. Chen, H. Qu, Z. Luo, S. Wu, H. A. Colorado, S. Wei and Z. Guo, Energy Environ. Sci., 2013, 6, 194–204 CAS.
- L. Borchardt, M. Oschatz and S. Kaskel, Mater. Horiz., 2014, 1, 157–168 RSC.
- O. Barbieri, M. Hahn, A. Herzog and R. Kötz, Carbon, 2005, 43, 1303–1310 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- E. Raymundo-Piñero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2006, 44, 2498–2507 CrossRef.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Béguin, Carbon, 2005, 43, 1293–1302 CrossRef CAS.
- ISO 9277:2010, Determination of the Specific Surface Area of Solids by Gas Adsorption—BET Method, ISO, 2010 Search PubMed.
- F. Stoeckli, M. V. López-Ramón, D. Hugi-Cleary and A. Guillot, Carbon, 2001, 39, 1115–1116 CrossRef CAS.
- D. Lozano-Castelló, M. A. Lillo-Ródenas, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2001, 39, 741–749 CrossRef.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- L.-C. Dolores, S.-G. Fabián, C.-A. Diego and L.-S. Ángel, in Carbons for Electrochemical Energy Storage and Conversion Systems, ed. F. Béguin and E. Frackowiak, CRC Press, 2009, pp. 115–162 Search PubMed.
- J. Romanos, M. Beckner, T. Rash, L. Firlej, B. Kuchta, P. Yu, G. Suppes, C. Wexler and P. Pfeifer, Nanotechnology, 2012, 23, 015401–015407 CrossRef CAS PubMed.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- A. Burke, J. Power Sources, 2000, 91, 37–50 CrossRef CAS.
- A. Burke and M. Miller, J. Power Sources, 2011, 196, 514–522 CrossRef CAS.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2013, 46, 1094–1103 CrossRef CAS PubMed.
- H. Shi, Electrochim. Acta, 1996, 41, 1633–1639 CrossRef CAS.
- D. Qu and H. Shi, J. Power Sources, 1998, 74, 99–107 CrossRef CAS.
- P. Azaïs, in Supercapacitors: Materials, Systems, and Applications, ed. F. Béguin and E. Frąckowiak, Wiley-VCH Verlag GmbH & Co., 2013, pp. 307–371 Search PubMed.
- L. Eliad, E. Pollak, N. Levy, G. Salitra, A. Soffer and D. Aurbach, Appl. Phys. A: Mater. Sci. Process., 2006, 82, 607–613 CrossRef CAS.
- E. Avraham, B. Yaniv, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2008, 112, 7385–7389 CAS.
- M. Noked, E. Avraham, Y. Bohadana, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2009, 113, 7316–7321 CAS.
- B. Xu, S. Yue, Z. Sui, X. Zhang, S. Hou, G. Cao and Y. Yang, Energy Environ. Sci., 2011, 4, 2826–2830 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of Cm values calculated from voltage ranges of 0 to 1 V, 0 to 0.9 V and 0 to 0.8 V; test set-up for measuring electrical conductivity; TGA of HTC-GA and HTC-G, N2 adsorption–desorption isotherms for AC-GFe-800-2h and AC-GFe-800-4h; XPS wide spectra and high-resolved C 1s spectra; relationship between the activation temperature and bulk elemental composition of ACs; XRD patterns; galvanostatic charge–discharge curves; CV curves at different scan rates; electrical conductivity of AC powders; cycling life time; Raman spectra; SEM, TEM, HRTEM, BF-STEM and HAADF-STEM images; EELS. See DOI: 10.1039/c6ra24398c |
| This journal is © The Royal Society of Chemistry 2016 |