
Microbial transformation of methyl cyperenoate by Cunninghamella elegans AS 3.2028 and the antithrombotic activities of its metabolites†
Jin-Long Tianab,
Yu Chenc,
Yu-Xi Wangab,
Xiao-Xiao Huangab,
Xue Sunab,
Ke-Chun Liud and
Shao-Jiang Song*ab
aSchool of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China. E-mail: songsj99@163.com; Fax: +86-24-23986088; Tel: +86-24-23986510
bKey Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China
cSchool of Life Science and Biopharmaceutics, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenyang 110016, People's Republic of China
dBiology Institute of Shandong Academy of Sciences, Jinan, China
First published on 24th November 2016
Abstract
Microbial transformation is a remarkable tool for the structural modification of bioactive natural compounds converting them into more valuable biologically active derivatives. In order to obtain the bioactive metabolites of methyl cyperenoate, it was biotransformed using Cunninghamella elegans AS 3.2028 to afford eight new derivatives (1–8) including six new hydroxylated metabolites (2–6, 8), one new oxidation metabolite (7), along with one unusual new dehydration metabolite (1). Metabolite 1 and metabolite 7 with unsaturated ring C were found in patchoulane sesquiterpene for the first time. In addition, the biotransformation time-course of methyl cyperenoate by Cunninghamella elegans AS 3.2028 was described and the possible transformation pathway was also proposed on the basis of structural analyses and biotransformation time-courses. It was noteworthy that the major biotransformation reactions only occurred in ring C of methyl cyperenoate. All the biotransformation metabolites were tested in vitro for antithrombotic activity by measuring the platelet count, together with the partial thromboplastin time (APTT), the thrombin time (TT) and the prothrombin time (PT). The biotransformation derivatives 1–3 exhibited potent antithrombotic activity in vitro. Furthermore, the active compounds 1–3 were tested for their in vivo antithrombotic activity using a transgenic zebrafish model and compound 1 exhibited good antithrombotic activity in this model. Interestingly, transformation derivative 1 displayed potent antithrombotic activity both in vivo and in vitro compared with its precursor (the original compound), suggesting that it could be a promising agent for further preclinical evaluation for preventing abnormal blood clotting.
Introduction
Thrombus formation is a crucial event in the pathophysiology of atherosclerotic cardiovascular diseases (CVDs). The leading causes of death (30% of the total) in the world are diseases involving the heart and blood vessels and, consequently, thrombosis.1–3 Platelet activation in atherosclerotic arteries is central to the development of arterial thrombosis and, therefore, a precise control of platelet function is imperative for preventing thrombotic events.4,5 So, research on novel bioactive compounds and drugs with different mechanisms of action, increased efficacy, and low toxicity is urgently needed. Owing to their huge chemical diversity, plant-derived natural products have emerged as one of the most promising ways to discover new agents.6,7 However, recently, natural product-based drug discovery has declined, partly due to the difficulty in isolating or synthesizing natural products and their analogues.8 Therefore, to retain their usefulness, it is important to construct structurally diverse compounds from natural products. Biotransformation entails using biological systems or interrelated enzymes of biological systems to modify natural compounds9–12 and is regarded of as an efficient and important tool to diversify structures, especially for complex natural products, which are difficult to modify and prepare by chemical approaches. Compared with chemical synthesis, microbial transformations are usually used as an alternative method to modify complex structures using multiple enzymatic reactions.13,14Methyl cyperenoate (MC) is one of the bioactive sesquiterpenes of Croton crassifolius, a plant used in traditional Chinese medicine to treat snake bite, stomach ache, sternalgia, joint pain, pharyngitis, jaundice, and rheumatoid arthritis in China and is used to treat cancer by the indigenous people in Thailand.15–18 Previous pharmacological studies have shown that MC has antimalarial,19 antifungal,20 and gastroprotective activity.21
Currently, great attention is being paid to the antithrombotic activity of sesquiterpenes.22–24 Our previous anti-platelet aggregation test demonstrated that MC exhibits anti-platelet aggregation activity. As part of our ongoing studies on antithrombotic terpenoids from herbal medicines, we have concentrated on MC and its structural analogues as an ideal target for the study of anti-platelet aggregation activity. Their structural modification is thus important for further evaluation of their antithrombotic activity. However, the great stability of the patchoulane skeleton makes structural modification of MC very difficult. Conventional chemical synthesis methods can only modify the position of the carbonyl group or double bond.21 To overcome these difficulties, we have developed a biotransformation method trying to acquire derivations of MC and shown more valuable biological activity.
In our research, methyl cyperenoate (MC) was transformed by Cunninghamella elegans AS 3.2028 for the first time. Six new hydroxylated metabolites (2–6, 8), one new oxidation metabolite (7), along with one unusual new dehydration metabolite (1) were isolated and identified. To the best of our knowledge, ring C of the tricyclic patchoulane-type sesquiterpenes is always saturated. Metabolite 7 (carbonylation at C-8) and metabolite 1 (with an unusual α,β-unsaturated ketone at C-8,9,10) were found with an unsaturated ring C for the first time. All biotransformed derivatives were tested for in vitro antithrombotic activity using a platelet count investigation. In addition, we examined their effects on the partial thromboplastin time (APTT), the thrombin time (TT) and the prothrombin time (PT) in a series of in vitro assays. Furthermore, the active metabolites 1–3 were tested for their in vivo antithrombotic activity using a transgenic zebrafish model.
Results and discussion
Identification of transformed products
Compound 1 was obtained as pale yellow oil. Its HR-ESI-MS gave a pseudo-molecular ion peak at m/z 283.1359 [M + Na]+, (calcd for C16H20O3Na 283.1310) indicating a molecular formula of C16H20O3. The 1H-NMR spectrum of 1 revealed some signals common to patchoulane-type sesquiterpenes, namely signals of two geminal methyl groups δH 0.99 (3H, s, H-12), and δH 0.98 (3H, s, H-13) and one methyl group signal δH 2.14 (3H, d, H-14) (Table 1). Compared with MC, the major differences were the presence of one carbonyl carbon at δC 202.9, two olefinic carbons at δC 166.0 and 123.7, and the absence of one methine (δC 35.9 in MC) and two methylene (δC 27.9 and 27.0 in MC) (Table 2) groups indicating that 1 was the hydroxylated-dehydration derivative of MC. The correlation from δH 5.85 (H-9) to δC 166.0 (C-10), and from δH 2.14 (H-14) to δC 166.0 (C-10)/δC 123.7 (C-9) in HMBC spectrum (Fig. 2) suggested a double bond at C-9 and C-10. The correlation from δH 5.85 (H-9)/δH 2.64 (H-7) to δC 202.9 (C-8), suggested the carbonyl was located at C-8, so these HMBC correlations established an unusual α,β-unsaturated ketone in ring C, as shown in Fig. 1. Therefore, compound 1 was shown to be methyl cyperenoate-9-dien-8-one.| Position | Comp. MC | Comp. 1 | Comp. 2 | Comp. 3 | Comp. 4 | Comp. 5 | Comp. 6 | Comp. 7 | Comp. 8 |
|---|---|---|---|---|---|---|---|---|---|
| a Data were measured in CDCl3-d at 400 MHz. Data were measured in CD3OD-d4 at 400 MHz. | |||||||||
| 2 | 1.51 (1H, m) | 2.00 (1H, m) | 1.59 (1H, m) | 1.63 (1H, m) | 1.58 (1H, m) | 1.60 (1H, m) | 1.61 (1H, m) | 1.67 (1H, m) | 1.81 (1H, m) |
| 1.74 (1H, m) | 2.26 (1H, m) | 1.74 (1H, m) | 1.89 (1H, m) | 1.80 (1H, m) | 1.75 (1H, m) | 1.87 (1H, m) | 1.97 (1H, m) | 1.86 (1H, m) | |
| 3 | 2.66 (1H, m) | 2.96 (1H, m) | 1.81 (1H, m) | 2.66 (1H, m) | 2.70 (1H, m) | 1.43 (1H, m) | 2.60 (1H, m) | 2.81 (1H, m) | 2.83 (2H, m) |
| 2.76 (1H, m) | 3.06 (1H, m) | 1.92 (1H, m) | 2.76 (1H, m) | 2.80 (1H, m) | 1.61 (1H, m) | 2.69 (1H, m) | 2.90 (1H, m) | ||
| 6 | 2.24 (1H, m) | 2.24 (1H, m) | 2.23 (1H, m) | 2.22 (1H, m) | 2.48 (1H, m) | 2.04 (1H, m) | 2.54 (2H, s) | 2.48 (1H, m) | 2.28 (1H, m) |
| 2.81 (1H, m) | 3.03 (1H, m) | 2.70 (1H, m) | 2.70 (1H, m) | 2.52 (1H, m) | 2.68 (1H, m) | 2.86 (1H, m) | 2.70 (1H, m) | ||
| 7 | 1.96 (1H, m) | 2.64 (2H, dd) | 2.04 (1H, m) | 2.15 (1H, m) | 2.00 (1H, m) | 2.10 (1H, m) | 2.46 (1H, m) | 2.00 (1H, m) | |
| 8 | 1.36 (1H, m) | 2.71 (1H, m) | 1.16 (1H, m) | 4.11 (1H, m) | 3.96 (1H, m) | 1.48 (1H, m) | 1.41 (1H, m) | ||
| 2.01 (1H, m) | 2.82 (1H, m) | 1.60 (1H, m) | 2.00 (1H, m) | 2.2 (1H, m) | |||||
| 9 | 1.14 (1H, m) | 5.85 (1H, s) | 3.34 (1H, m) | 1.38 (1H, m) | 1.85 (1H, m) | 2.71 (1H, m) | 1.08 (1H, m) | 2.12 (1H, m) | 1.54 (1H, m) |
| 1.47 (1H, m) | 1.93 (1H, m) | 2.82 (1H, m) | 1.66 (1H, m) | 2.29 (1H, m) | 1.67 (1H, m) | ||||
| 10 | 2.06 (1H, m) | 1.96 (1H, m) | 2.13 (1H, m) | 2.09 (1H, m) | 2.33 (1H, m) | 2.08 (1H, m) | 2.31 (1H, m) | ||
| 12 | 0.82 (3H, s) | 0.99 (3H, s) | 0.83 (3H, d) | 0.92 (3H, s) | 0.88 (3H, s) | 0.82 (3H, s) | 0.84 (3H, s) | 0.96 (3H, s) | 0.81 (3H, s) |
| 13 | 0.99 (3H, s) | 0.98 (3H, s) | 1.04 (3H, s) | 3.76 (3H, s) | 1.00 (3H, s) | 1.25 (3H, s) | 0.94 (3H, s) | 0.91 (3H, s) | 1.25 (3H, s) |
| 14 | 0.85 (3H, d) | 2.14 (3H, d) | 1.05 (3H, d) | 0.87 (3H, d) | 0.90 (3H, s) | 0.89 (3H, d) | 0.85 (3H, d) | 1.04 (3H, d) | 1.19 (3H, s) |
| OCH3 | 3.74 (3H, s) | 3.73 (3H, s) | 3.72 (3H, s) | 3.72 (3H, s) | 3.73 (3H, s) | 3.72 (3H, s) | 3.72 (3H, s) | 3.74 (3H, s) | 3.73 (3H, s) |
| Position | Comp. MC | Comp. 1 | Comp. 2 | Comp. 3 | Comp. 4 | Comp. 5 | Comp. 6 | Comp. 7 | Comp. 8 |
|---|---|---|---|---|---|---|---|---|---|
| a Data were measured in CDCl3-d at 100 MHz. Data were measured in CD3OD-d4 at 100 MHz. | |||||||||
| 1 | 67.8 | 72.2 | 66.9 | 67.2 | 66.8 | 67.5 | 68.2 | 67.8 | 71.0 |
| 2 | 25.7 | 22.1 | 25.9 | 26.6 | 24.5 | 25.6 | 26.1 | 24.9 | 22.9 |
| 3 | 36.7 | 37.1 | 36.7 | 36.5 | 36.4 | 36.7 | 34.4 | 36.7 | 37.2 |
| 4 | 123.3 | 124.6 | 124.0 | 123.3 | 123.4 | 124.6 | 123.7 | 125.8 | 124.1 |
| 5 | 169.5 | 165.4 | 167.9 | 169.0 | 168.6 | 167.6 | 165.1 | 165.6 | 169.3 |
| 6 | 31.1 | 27.6 | 31.6 | 30.7 | 25.0 | 31.3 | 38.5 | 29.6 | 30.0 |
| 7 | 48.2 | 64.6 | 48.0 | 45.3 | 54.5 | 54.3 | 81.8 | 64.2 | 47.7 |
| 8 | 27.0 | 202.9 | 36.8 | 27.7 | 66.5 | 73.4 | 34.0 | 213.9 | 24.9 |
| 9 | 27.9 | 123.7 | 71.7 | 26.0 | 36.5 | 36.6 | 28.8 | 43.0 | 33.7 |
| 10 | 35.9 | 166.0 | 44.2 | 35.2 | 34.8 | 33.6 | 34.9 | 36.3 | 77.5 |
| 11 | 41.6 | 48.5 | 41.6 | 45.7 | 41.6 | 41.5 | 45.6 | 43.7 | 42.3 |
| 12 | 26.2 | 17.6 | 25.7 | 19.7 | 25.4 | 27.2 | 19.1 | 24.1 | 27.7 |
| 13 | 19.3 | 23.5 | 25.7 | 63.7 | 17.7 | 20.8 | 15.3 | 18.6 | 22.1 |
| 14 | 18.0 | 18.0 | 13.9 | 17.0 | 16.7 | 17.7 | 16.7 | 16.3 | 28.7 |
| 15 | 166.1 | 165.3 | 165.9 | 166.2 | 166.1 | 165.9 | 166.0 | 165.6 | 166.1 |
| –OCH3 | 51.0 | 50.5 | 51.1 | 50.2 | 50.2 | 51.1 | 50.2 | 50.5 | 51.1 |
 | ||
| Fig. 1 Structures of biotransformed products of methyl cyperenoate (MC) by C. elegans AS 3.2028. | ||
 | ||
| Fig. 2 Key HMBC correlations (C–H) of 1–8. | ||
Compound 2 gave a pseudo-molecular ion peak at m/z 287.1617 [M + Na]+ by HR-ESI-MS, (calcd for C16H24O3Na 287.1623) indicating a molecular formula of C16H24O3. The 1H-NMR and 13C NMR spectral data were similar to that of MC, suggesting that compound 2 had the same scaffold as MC (Tables 1 and 2). The major differences involved the presence of one oxygenated methine group (δC 71.7 in 2) and the absence of one methylene (δC 27.9 in MC) indicating that 2 was the hydroxylated derivative of MC. The HMBC correlation (Fig. 2) from δH 1.05 (H-14)/δH 1.96 (H-10)/δH 2.04 (H-7)/[δH 2.71 (H-8), 2.82 (H-8)] to δC 71.7 (C-9) suggested that the hydroxyl group was located at C-9. The relative configuration of 2 was established by a NOESY experiment. In the NOE spectrum (Fig. 3), the signal between H-14 (δH 1.05) and H-9 (δH 3.34) showed that the groups were on the same face and arbitrarily assigned as β. Therefore, compound 2 was identified as 9β-hydroxy-methyl cyperenoate.
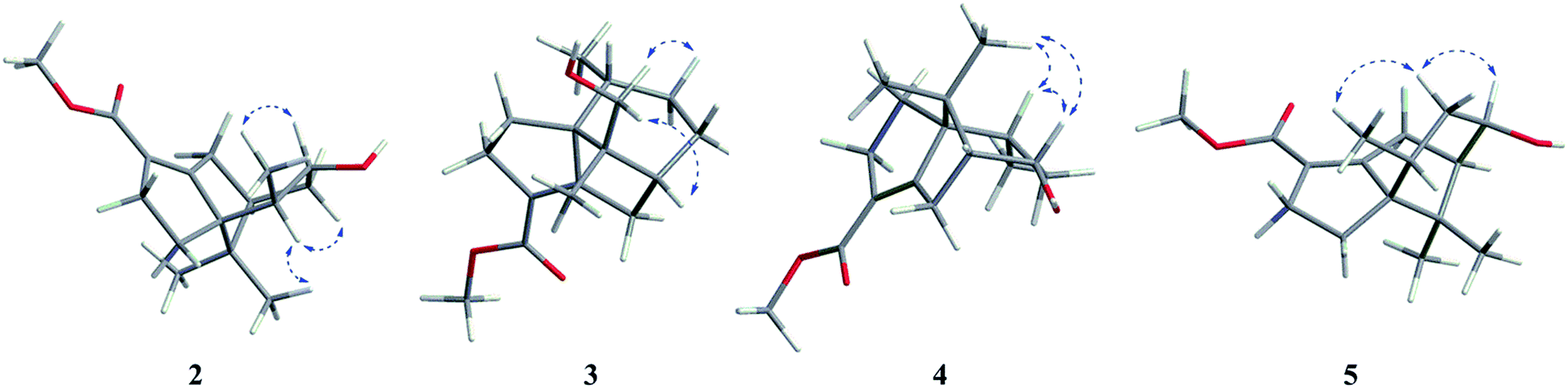 | ||
| Fig. 3 Key NOESY correlations (H–H) of 2–5. | ||
Compound 3 had the molecular formula C16H24O3 (HR-ESI-MS at m/z 287.1617 [M + Na]+, calcd for C16H24O3Na 287.1623). Comparison of the 13C NMR data of 3 with those of MC showed the presence of one oxygenated methylene group (δC 63.7 in 3) and the absence of one methyl (δC 26.2 in MC), indicating that 3 was the hydroxylated derivative of MC (Table 2). Their locations were confirmed by HMBC correlations (Fig. 2) from δH 3.76 (H-13) to δC 67.2 (C-1)/δC 19.7 (C-13)/δC 45.7 (C-11)/δC 45.3 (C-7), which suggested that the hydroxyl group was located at C-13. Thus, compound 3 was identified as 13-hydroxy-methyl cyperenoate.
Compound 4 was assigned the molecular formula C16H24O3 on the basis of its HRESIMS (m/z 287.1618 [M + Na]+, calcd for C16H24O3Na 287.1623). Comparison of the 13C NMR spectrum of 4 with that of MC (Table 2), showed that there was one oxygenated methine group at δC 66.5 in 4, while the methylene at δC 27.0 in MC disappeared indicating that 4 was the hydroxylated derivative of MC. The correlation from δH 1.85 (H-9)/δH 2.00 (H-7)/δH 2.48 (H-6) to δC 66.5 (C-8) in the HMBC spectrum (Fig. 2) suggested that the hydroxyl group was located at C-8. In the NOE spectrum (Fig. 3), the signal between H-8 (δH 4.11) and H-13 (δH 1.00)/H-10 (δH 2.09) confirmed that the groups were on the same face and arbitrarily assigned as α. Compound 4 was identified as 8α-hydroxy-methyl cyperenoate.
Compound 5 had the molecular formula C16H24O3, as indicated by the HRESIMS at m/z 287.1604 [M + Na]+, (calcd for C16H24O3Na 287.1623). Comparison of the 1H and 13C NMR data of 5 with those of MC (Tables 1 and 2), showed the presence of one oxygenated methine group (δC 73.4 in 8) and the absence of one methylene (δC 27.0 in MC) indicating that 5 was the hydroxylated derivative of MC. The correlation from δH 2.10 (H-7)/δH 2.71 (H-9)/δH 2.33 (H-10) to δC 73.4 (C-8) in the HMBC spectrum (Fig. 2) suggested that the hydroxyl group was located at C-8. The signal between H-8 (δH 3.96) and H-9 (δH 2.71), and H-9 (δH 2.71) and H-10 (δH 2.33) in the NOESY correlations confirmed that the groups were on the same face and arbitrarily assigned as β (Fig. 3). Compound 5 was identified as 8β-hydroxy methyl-cyperenoate.
The molecular formula of metabolite 6 was C16H24O3 from the positive-ion HRESIMS ([M + Na]+ at m/z 287.1638, calcd for C16H24O3Na 287.1623) in combination with the 13C NMR spectroscopic data (Table 2). The major differences between MC were the presence of one oxygenated quaternary carbon (δC 81.8 in 6) and the absence of one methylene (δC 48.2 in MC) indicating that 6 was the hydroxylated derivative of MC. The correlation from δH [1.48 (H-8) and δH 2.00 (H-8)]/δH 1.66 (H-9)/δH 2.54 (H-6) to δC 81.8 (C-7) in HMBC spectrum (Fig. 2) suggested that the hydroxyl group was located at C-7. Compound 6 was identified as 7-hydroxy-methyl cyperenoate.
Metabolite 7 was assigned the molecular formula C16H22O3 according to the HRESIMS (m/z 285.1547 [M + Na]+, calcd for C16H24O3Na 285.1467). Analyses of the spectroscopic data suggested that 7 was a ketone carbonylation derivative of MC (Tables 1 and 2), from the presence of one carbonyl carbon at δC 213.9 and the absence of one methylene (δC 27.0) in MC. The correlation from δH 2.31 (H-10)/δH 2.12 (H-9)/δH 2.46 (H-7) to δC 213.9 (C-8) in the HMBC spectrum (Fig. 2) suggested that the carbonyl group was located at C-8. Compound 7 was identified as methyl cyperenoate-8-one.
Metabolite 8 was also a hydroxylated derivative of MC, as deduced by the HRESIMS (m/z 285.1547 [M + Na]+), (calcd for C16H24O3Na 285.1467) and NMR data (Tables 1 and 2). The correlation from δH 1.19 (H-9)/δH 1.54 (H-9)/δH 1.41 (H-8) to δC 77.5 (C-10) in HMBC spectrum (Fig. 2) suggested that the hydroxyl group was located at C-10 and the change in H-6 from a doublet to a singlet, also suggested that the hydroxyl was located at C-10. Therefore, compound 8 was identified as 10-hydroxy-methyl cyperenoate.
Biotransformation time-course
The biotransformation time-course of MC by C. elegans AS 3.2028 was investigated by us (Fig. 4). Cultures of C. elegans AS 3.2028 incubated with MC were examined by HPLC after an incubation period of 48 h (Fig. 5). The substrate MC was almost completely exhausted within 48 h to produce metabolites with compound 3 as the major metabolite (yield 19.1%) and 13-hydroxymethyl cyperenoate (3) was a hydroxylated derivative of MC. The amounts of metabolites 4 and 5 decreased progressively from 18 h to 48 h (Fig. S63†), suggesting that they were further transformed to produce other metabolites, such as 1 (hydroxylated-dehydration metabolite) or 7 (hydroxylated-oxygenation metabolite). Therefore, it would appear that metabolites 4 and 5 were important intermediates in the metabolic progress of MC by C. elegans AS 3.2028. The time-courses of metabolites 2, 3, 6 and 8 (hydroxylated derivatives) indicated that the highest amounts were obtained at 36 h. Thus, the biotransformation time-course suggested that C. elegans AS 3.2028 transformed MC by hydroxylation (2–6 and 8), oxygenation (8) and dehydration (1). | ||
| Fig. 4 Time courses of biotransformation of MC by C. elegans AS 3.2028. | ||
 | ||
| Fig. 5 HPLC chromatograms of biotransformation of methyl cyperenoate (MC) by C. elegans AS 3.2028 after the incubation period of 48 h. | ||
Possible metabolic pathways are outlined in Fig. 1. Regio- and stereoselective hydroxylation at C-7, C-8, C-9, C-10 and C-13 yielded metabolites 2–6, hydroxylated-oxygenation product 7 and hydroxylated-dehydration derivative 1 during the biotransformation process, respectively. The formation of biotransformation metabolite 1 is proposed to involve hydroxylation, oxidation and unusual dehydration reactions. The possible metabolic pathways of metabolite 1 are listed in Fig. 6. Also the intermediate product of oxidation was isolated (metabolite 7) as part of the biotransformation of metabolites, and the biotransformation time-course was examined. This reaction is interesting because it permitted the formation of the unsaturated ring C of patchoulane sesquiterpene reported for the first time. It is noteworthy that the major biotransformation reactions only involve ring C of MC. This type of catalytic ability of C. elegans AS 3.2028 to cause hydroxylation oxygenation and degradation of patchoulane sesquiterpenoid deserves further investigation to enhance the chemical diversity of sesquiterpenoids.
 | ||
| Fig. 6 The proposed biotransformation pathways of biotransformed product 1. | ||
Antithrombotic assay
 | ||
| Fig. 7 Antiplatelet aggregation activity of MC and biotransformation derivatives 1–8 (X ± SD, n = 3). | ||
Anticoagulation assay
The pre-clinical evaluation of the antithrombotic potential of novel molecules requires the use of reliable and reproducible experimental models. PT, APTT and TT are the most established and commonly used preparations to determine the efficacy of novel antithrombotic drugs.26–28 Therefore, the anticoagulant activity of MC and biotransformation derivatives 1–3 were tested using APTT, TT and PT assays. Although the anticoagulant activity of biotransformation derivatives 1–3 were weaker than that of aspirin, the APTT, TT and PT were significantly prolonged by biotransformation derivatives 1–3 (10 μM). A prolongation of the APTT suggests inhibition of the intrinsic and/or the common pathway, while a PT and TT prolongation suggests inhibition of the extrinsic and/or the common pathway.25 Therefore, our results indicate that biotransformation derivatives 1–3 can inhibit the common coagulation pathway. From the results of the two antithrombotic evaluation models in vitro, the activities of the biotransformation products had been significantly improved compared with their precursor (MC) (Fig. 7 and Table 3). A simple structure–activity relationship analysis was carried out on the biotransformation products, and we found that the hydroxylation at C-9 and C-13 produced better antithrombotic activity. It was noteworthy that the double bond at ring C (C-9 and C-10) of derivative 1 had significant effect on the antithrombotic activity in vitro, as derivative 7 (no double bond at ring C) did not show any significant antithrombotic activity.| Sample | Dose | APTT (s) | TT (s) | PT (s) | PT (INR) |
|---|---|---|---|---|---|
| a All data are expressed as the means ± SD of three independent experiments. *p < 0.05, **p < 0.01 compared with the control group. | |||||
| Control | Saline | 24.8 ± 1.0 | 14.2 ± 0.5 | 11.2 ± 0.5 | 1 |
| MC | 10 μM | 25.9 ± 0.5 | 14.9 ± 0.4 | 11.7 ± 0.4 | 1.04 |
| 1 | 10 μM | 35.7 ± 1.4** | 20.9 ± 2.0 | 14.9 ± 0.3* | 1.33 |
| 2 | 10 μM | 31.4 ± 0.9** | 17.9 ± 0.4* | 14.0 ± 0.2* | 1.25 |
| 3 | 10 μM | 31.3 ± 0.6** | 17.9 ± 1.0* | 14.8 ± 0.5* | 1.32 |
| Aspirin | 10 μM | 53.1 ± 1.8** | 27.2 ± 1.4** | 23.1 ± 1.1** | 2.06 |
Growth inhibition effects of biotransformation derivatives 1–3 against peripheral blood lymphocytes cells
In order to ascertain the cytotoxicity of 1–3 on peripheral blood lymphocytes, rat peripheral blood lymphocytes cells were prepared and treated with 0, 5, 10, 20, 40 and 80 μM compounds 1–3 for 48 h. Also, the cells were treated with the same doses of aspirin as positive controls. The results showed that the cell viability of peripheral blood lymphocytes cells treated with 1–3 were no different with aspirin at all tests doses and exhibited high cell viability (Fig. 8), indicating that the compounds 1–3 exhibited no in vitro cellular cytotoxicity. | ||
| Fig. 8 The growth inhibitory activity of 1–3 in peripheral blood lymphocytes cells, compared with aspirin were measured using an MTT assay. The peripheral blood lymphocytes cells were treated with 1–3 and aspirin at 0–80 μM for 48 h (means ± SD of three independent experiments). | ||
Antithrombotic assay using a transgenic zebrafish system
To confirm these in vitro results, the in vivo antithrombotic activity in the transgenic zebrafish model was determined. In this study, the antithrombotic activity was induced by FeCl3 treatment in vivo, while biotransformation derivatives 1–3 inhibited the thrombus production induced by FeCl3 (Table 4).3 Among these compounds, 1 showed significant antithrombotic activity compared with the control group (4.40 ± 0.42 min), and the time to form a thrombus in the tested zebrafish was clearly prolonged (6.56 ± 0.35 min). So, compound 1 exhibited its antithrombotic activity in this zebrafish model.| Samples | Concentration | Thrombus formation time (min) |
|---|---|---|
| a All data are expressed as the means ± SD of three independent experiments. *p < 0.05, **p < 0.01 compared with the control group. | ||
| Control | 0.1% DMSO | 4.40 ± 0.42 |
| Heparin | 20 U mL | 8.40 ± 0.32* |
| MC | 1 μg mL−1 | 4.70 ± 0.52 |
| 10 μg mL−1 | 4.20 ± 0.26 | |
| 50 μg mL−1 | 4.12 ± 0.36 | |
| 1 | 1 μg mL−1 | 4.50 ± 0.53 |
| 10 μg mL−1 | 4.90 ± 0.48 | |
| 100 μg mL−1 | 6.56 ± 0.35* | |
| 2 | 1 μg mL−1 | 4.20 ± 0.49 |
| 10 μg mL−1 | 4.43 ± 0.55 | |
| 100 μg mL−1 | 5.20 ± 0.47 | |
| 3 | 1 μg mL−1 | 4.10 ± 0.33 |
| 10 μg mL−1 | 4.30 ± 0.55 | |
| 100 μg mL−1 | 5.20 ± 0.49 | |
The results of the antithrombotic testing showed that biotransformation derivatives 1–3 exhibited potent antiplatelet aggregation activity in rat plasma, suggesting that these active compounds could be an interesting alternative acting on the ADP pathway. Also, APTT, TT and PT were significantly prolonged by biotransformation derivatives 1–3, in agreement with the platelet count results. The consistency of the experimental data of the two groups in vitro showed that the results of the antithrombotic assay were objective and reliable. However, the data presented showed that only the hydroxylated-dehydration derivative 1 exhibited significant activity in vivo, while hydroxylated derivatives 2 and 3 exhibited no activity. This phenomenon might be due differences in bioactivity between in vivo and in vitro conditions. Note that, through the process of biotransformation, we found one unusual dehydration new metabolite 1, which exhibited significant antithrombotic and antiplatelet aggregation activity than that of its precursor (MC).
The possible action mechanism of the active compounds was explored by using molecular docking studies. Through the virtual screening of a large number of anti-thrombotic targets, we found that compound 1 had better ligand–receptor interaction with eNOS (Fig. S64†). Based on the molecular docking studies, we can speculate that the compound 1 may be an activator of eNOS which deserves further research.
Experimental
General experimental procedures
1D and 2D-NMR were recorded in methanol-d4 and chloroform-d by Bruker-400 spectrometer (400 MHz for 1H-NMR and 100 MHz for 13C-NMR). HR-ESI-TOF/MS experiments were carried out on a MicroTOF spectrometer (Bruker Co., Karlsruhe, Germany). Analytical liquid chromatography was performed on an Agilent 1200 HPLC instrument equipped with photodiode array detector (PAD). Preparative HPLC was performed on a Shimadzu instrument with an UV detector and a YMC ODS A column (250 × 20 mm, 5 mm). Column chromatography was performed with silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, People's Republic of China). TLC was carried out on glass pre-coated silica gel GF254 plates. Spots were visualized under UV light or by spraying with 10% sulfuric acid in EtOH followed by heating. All solvents including ethyl acetate, methanol, CHCl3 and acetone are A.R. grade and were purchased from Tianjin Damao Chemical Reagent Company (Tianjing, China). Methanol and acetonitrile for HPLC analysis are chromatographic grade (YuWang, TianJin, China).Plant material
The roots of C. crassifolius were collected Guangxi Province, China, in June 2014. Samples were authenticated by Prof. Jin-Cai Lu from the Department of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University. The voucher specimen (no. 20140711) is lodged in the herbarium of Shenyang Pharmaceutical University, Liaoning, China.Microorganisms and media
C. elegans AS 3.2028 was obtained from the China General Microbiological Culture Collection Center in Beijing, P. R. China and maintained on potato dextrose agar slants, which were produced using the following procedure: 200 g minced, peeled potato was boiled in distilled water for 1 h, and then the extract was filtered. The filtrate was then diluted with distilled water to 1 L, and 20 g glucose and 15 g agar were added. The biotransformation medium was composed of polypeptone (5 g L−1), sucrose (15 g L−1), glucose (15 g L−1), K2HPO4 (1 g L−1), FeSO4·7H2O (0.01 g L−1), MgSO4·7H2O (0.5 g L−1) and KCl (0.5 g L−1) in distilled water, with a pH of 7.0.Biotransformation procedure
A spore suspension of C. elegans AS 3.2028 was first prepared and suitably diluted and then inoculated into a 250 mL Erlenmeyer flask containing 50 mL of liquid medium at 28 °C on a rotary shaker (220 rpm) for 48 h. After 48 h of pre-culture, 5 mL aliquots of pre-culture were transferred into Erlenmeyer flasks (250 mL) containing 50 mL of medium, and the substrates were dissolved in ethanol at a concentration of 25 mg mL−1. An ethanol solution of MC was then added to the flask to achieve a final concentration of 0.2 mg mL−1 and the cultures were incubated for 48 h. Culture controls consisted of fermentation medium in which fungi were grown in the presence of the same amount of ethanol but in the absence of substrate. Substrate controls consisted of medium and the same concentration of substrate without the strain. Culture and substrate controls were performed in parallel with the biotransformation experiments. Preparative scale biotransformations of MC by C. elegans AS 3.2028 were performed in 500 mL Erlenmeyer flasks containing 100 mL pre-cultured medium. One hundred milligrams of MC were dissolved in 4 mL ethanol and added to each flask to achieve a final concentration of 0.5 mg mL−1. In total, 500 mg trantinterol was used to prepare the products. The other procedures were the same as those used in the biotransformation above.Extraction, purification and identification of biotransformation products
The dried and powdered roots of C. crassifolius (5.0 kg) were percolated with 75% EtOH at room temperature. Removal of EtOH from the extract under reduced pressure yielded a dark residue (1208 g), which was then suspended in H2O and extracted with petroleum ether, EtOAc, and n-butanol. The EtOAc extract was chromatographed on a silica gel column with gradient mixtures of petroleum ether–acetone (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 100:100). Seven fractions were collected and examined by TLC on silica gel. Fraction 2 was crystallized from hexane–acetone to yield MC (370 mg).
:1 → 100:100). Seven fractions were collected and examined by TLC on silica gel. Fraction 2 was crystallized from hexane–acetone to yield MC (370 mg).
The medium containing MC was filtered under vacuum, and the filtrate was extracted three times with an equivalent volume of ethyl acetate saturated with water. The organic phase was collected and concentrated in vacuo at 50 °C. The concentrated crude ethyl acetate extract was further separated by RP-C18 silica gel column chromatography (MeOH/H2O 40:60 → 100:0, v/v) to provide two fractions (Frs. 1–2), followed by preparative HPLC (70% MeOH/H2O) to yield compounds 1 (8.6 mg), 2 (6.2 mg), 3 (5.4 mg), 4 (7.7 mg), 5 (9.1 mg), 6 (5.8 mg), 7 (6.9 mg), 8 (4.5 mg).
HPLC analysis
The samples were analyzed on an Agilent 1260 equipped with a diode array detector at 240 nm and an Agilent Zorbax SB-C18 column, 4.6 mm × 250 mm (5 μm). Mobile phase was MeOH–H2O (0.05% TFA) (40:60 (v/v)) for 15 min followed by a linear gradient to (70:30 (v/v)) within 35 min and held for an additional 10 min. The flow rate was 0.8 mL min−1, and column temperature was set at 30 °C.
Platelet counting
Evaluation of the antiplatelet aggregation activities of the different samples was carried out using an ethanol extract, a 55% ethanol elution fraction, pure compounds 1–8, positive control (aspirin) and a blank (ethanol). Stock solutions (50 mg mL−1) of all samples were prepared by dissolving the test compounds in 100% DMSO. A rat was sacrificed to obtain platelet-rich plasma (PRP). This involved mixing fresh rat blood with 38 g L−1 sodium citrate (9:1, v/v) and then centrifuging the mixture for 10 min at 1500 × g. The supernatant obtained was used as PRP. PRP was prewarmed at 37 °C and then secondary metabolites were dissolved in 5% dimethyl sulphoxide (DMSO) at a concentration of 1 mg mL−1 (50 μL) and added to 450 μL samples of PRP, and incubated for 5 min at 37 °C. Then, 20 μL ADP (10 mol L−1) was added to the PRP to induce acute thrombosis. In blank and positive control experiments, normal saline and aspirin were added instead of the test samples, respectively. The platelet aggregation rate in rat plasma was recorded after ADP was added. Each analyte was tested six times, and an average value was calculated. The antiplatelet activity was expressed as the percentage inhibition of the control value. The results obtained are shown in Fig. 7.
Anticoagulation assay
APTT (activated partial thromboplastin time), TT (thrombin time) and PT (prothrombin time) were determined using a Hemoglutination analyzer (Genius CA51, China), according to the manufacturer's instructions as described. In brief, citrated mouse plasma (20 μL) was mixed with 10 μL MC or compounds 1–3 and incubated for 1 min at 37 °C. APTT assay reagent (20 μL) was added followed by incubation for 1 min at 37 °C, and then 20 mM CaCl2 (20 μL) was added. Clotting times were recorded. For TT assays, citrated mouse plasma (30 μL) was mixed with 10 μL MC or compounds 1–3 and incubated for 1 min at 37 °C, and then the clotting time was recorded. For PT assays, citrated normal human plasma (90 μL) was mixed with 10 μL of PLT or OA stock and incubated for 1 min at 37 °C. PT assay reagent (200 μL), which has been preincubated for 10 min at 37 °C, was then added and clotting time was recorded. The PT results were expressed in seconds and as International Normalized Ratios (INR), and the TT and APTT results were expressed in seconds. INR = (PT sample/PT control).Preparation of peripheral blood lymphocytes cells
Fresh peripheral blood was collected from rats. The fresh blood samples were separated using 15 mL sterile conical centrifuge tubes. Ficoll–Hypaque solution was slowly layered underneath the blood/PBS mixture by placing the tip of the pipette containing the solution at the bottom of the sample tube; 3 mL of Ficoll–Hypaque solution per 10 mL of blood/PBS mixture was used. Then, this was centrifuged for 20 min at 400g. The peripheral blood lymphocytes cells were harvested by careful pipetting of the corresponding density band and washed in PBS (250g, 10 min) to remove platelets. The peripheral blood lymphocytes cells were resuspended in complete DMEM (10% NBCS), and the cells were counted and incubated in 96-well culture clusters, followed by an MTT assay. All experiments and procedures were carried out according to the Regulations of Experimental Animal Administration issued by State Committee of Science and Technology of China, and approved by the Institutional Ethical Committee (IEC) of Shenyang Pharmaceutical University.Antithrombotic assay using a transgenic zebrafish system
Four days post fertilization zebrafish (CD41:GFP) larvae were placed in a 24 well plate with six fish per well and one compound concentration (100 μg mL−1) per well. The zebrafish larvae were exposed to one concentration (100 μg mL−1) of the compound for 24 h while, we added a little DMSO (=500 μg L−1) as a cosolvent to improve the compound solubility. Then, the plate was exposed to 1 ppm FeCl3, and the time was recorded until a thrombus was observed in the venous system. Control group was the 0.1% DMSO and 20 U mL−1 heparin sodium set as the positive control group in this assay. The thrombosis time of the control group, positive control group and the biotransformation products were shown in Table 4.Structure characterization of new compounds
Conclusions
In this paper, biotransformation was used to catalyze the modification of methyl cyperenoate (MC) by C. elegans AS 3.2028 for the first time. Eight biotransformation derivatives, including six new hydroxylated metabolites (2–6, 8), one new oxidation metabolite (7), along with one unusual new dehydration metabolite (1), were isolated and identified. Metabolite 7 (carbonylation at C-8) and metabolite 1 (form an unusual α,β-unsaturated ketone at C-8,9,10) were found with an unsaturated ring C for the first time. In addition, the possible transformation pathway was proposed on the basis of structural analyses and biotransformation time-courses, suggesting that the unusual dehydration metabolite (1) may be a further transformation derivative from 4 or 5. It was noteworthy that the major biotransformation reactions only occur in ring C of MC. All biotransformation derivatives were tested for in vitro antithrombotic activity by a platelet count investigation, together with assays of the partial thromboplastin time (APTT), thrombin time (TT) and prothrombin time (PT) in vitro. In addition, compounds 1–3 exhibited no in vitro cellular cytotoxicity form the peripheral blood lymphocytes test. Furthermore, metabolites 1–3 were tested for their in vivo antithrombotic activity using a transgenic zebrafish model. The possible action mechanism of the compound 1 was explored by using molecular docking studies, and we found that compound 1 had better ligand–receptor interaction with eNOS which deserves further research. The experiment results obtained showed that compound 1 displayed potent anticoagulant activity both in vivo and in vitro, suggesting that it could be a promising agent for further preclinical evaluation to prevent abnormal blood clotting. Moreover, the antithrombotic activity of the biotransformation products is significantly higher than that of their precursor (MC), emphasizing the great potential of this biotransformation approach for identifying new drug leads.Acknowledgements
This work was supported by the Project of Innovation Team (LT2015027) of Liaoning of P. R. China.Notes and references
- A. J. Lorenzatti and B. M. Retzlaff, Int. J. Cardiol., 2016, 221, 581–586 CrossRef PubMed
.
- P. Zhu, W. W. Sun, C. L. Zhang, Z. Y. Song and S. Lin, Int. J. Cardiol., 2016, 220, 235–241 CrossRef PubMed
- L. Z. Li, P. Y. Gao, S. J. Song, Y. Q. Yuan, C. T. Liu, X. X. Huang and Q. B. Liu, J. Funct. Foods, 2015, 12, 237–245 CrossRef CAS
- V. B. Damodaran, V. Leszczak, K. A. Wold, S. M. Lantvit, K. C. Popat and M. M. Reynolds, RSC Adv., 2013, 3, 24406–24414 RSC
- D. Veach, H. Hosking, K. Thompson and A. B. Santhakumar, Food Funct., 2016, 7, 3609–3616 CAS
- J. Eder, R. Sedrani and C. Wiesmann, Nat. Rev. Drug Discovery, 2014, 13, 577–587 CrossRef CAS PubMed
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 3, 311–335 CrossRef PubMed
- J. W. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed
- M. Xu, X. K. Huo, X. G. Tian, P. P. Dong, C. Wang, S. S. Huang, B. J. Zhang, H. L. Zhang, S. Denga and X. C. Ma, RSC Adv., 2014, 4, 10627–10647 RSC
- W. F. Liang, Z. W. Li, S. Ji, Q. Wang, X. Qiao, D. A. Guo and M. Ye, RSC Adv., 2015, 5, 63753–63756 RSC
- Z. Shang, A. A. Salim, Z. Khalil, P. V. Bernhardt and R. J. Capon, J. Org. Chem., 2016, 81, 6186–6194 CrossRef CAS PubMed
- M. Heidary and Z. Habibi, J. Mol. Catal. B: Enzym., 2016, 126, 32–36 CrossRef CAS
- L. M. T. Frija, R. F. M. Frade and C. A. M. Afonso, Chem. Rev., 2011, 111, 4418–4452 CrossRef CAS PubMed
- K. B. Borges, W. D. S. Borges, R. D. Patrón, M. T. Pupo, P. S. Bonato and I. G. Collado, Tetrahedron: Asymmetry, 2009, 20, 385–397 CrossRef CAS
- Z. X. Zhang, H. H. Li, F. M. Qi, L. L. Dong, Y. Hai, G. X. Fan and D. Q. Fei, RSC Adv., 2014, 4, 30059–30061 RSC
- G. C. Wang, J. G. Li, G. Q. Li, J. J. Xu, X. Wu, W. C. Ye and Y. L. Li, J. Nat. Prod., 2012, 75, 2188–2192 CrossRef CAS PubMed
- M. S. Qiu, D. Cao, Y. H. Gao, S. H. Li, J. P. Zhu, B. Yang, L. Zhou, Y. Zhou, J. Jin and Z. X. Zhao, Fitoterapia, 2016, 108, 81–86 CrossRef CAS PubMed
- W. H. Huang, J. J. Wang, Y. Y. Liang, W. Ge, G. C. Wang, Y. L. Li and H. Y. Chung, J. Ethnopharmacol., 2015, 175, 185–191 CrossRef CAS PubMed
- V. Hadi, M. Hotard, T. Ling, Y. G. Salinas, G. Palacios, M. Connelly and F. Rivas, Eur. J. Med. Chem., 2013, 65, 376–380 CrossRef CAS PubMed
- H. Achenbacht and G. Benirschke, Phytochemistry, 1997, 45, 149–157 CrossRef
- M. Pertino, J. A. Rodríguez, C. Theoduloz, I. Razmilic and G. S. Hirschmann, J. Pharm. Pharmacol., 2006, 58, 1507–1513 CrossRef CAS PubMed
- Q. Xia, X. Wang, D. J. Xu, X. H. Chen and F. H. Chen, Thromb. Res., 2012, 130, 409–414 CrossRef CAS PubMed
- Y. Tao and Y. Wang, Fitoterapia, 2010, 81, 393–396 CrossRef CAS PubMed
- C. C. Zhou, X. X. Huang, P. Y. Gao, F. F. Li, D. M. Li, L. Z. Li and S. J. Song, J. Asian Nat. Prod. Res., 2014, 16, 169–174 CrossRef CAS PubMed
- N. Y. Yang, G. S. Zhou, Y. P. Tang, H. Yan, S. Guo, P. Liu, J. A. Duan, B. S. Song and Z. Q. He, Fitoterapia, 2011, 82, 692–695 CrossRef CAS PubMed
- L. Lei, Y. B. Xue, Z. Liu, S. S. Peng, Y. He, Y. Zhang, R. Fang, J. P. Wang, Z. W. Luo, G. M. Yao, J. W. Zhang, G. Zhang, H. P. Song and Y. H. Zhang, Sci. Rep., 2015, 5, 13544–13552 CrossRef CAS PubMed
- S. K. Ku, I. C. Lee, J. A. Kim and J. S. Bae, Fitoterapia, 2013, 91, 1–8 CrossRef CAS PubMed
- W. Lee and J. S. Bae, J. Funct. Foods, 2015, 17, 388–398 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR, HRESIMS, UV, IR, CD spectrum, table and figure. See DOI: 10.1039/c6ra24332k |
| This journal is © The Royal Society of Chemistry 2016 |