
Synthesis and application of PNP pincer ligands in rhodium-catalyzed hydroformylation of cycloolefins†
Qianhui Wua,
Fanding Zhoua,
Xiao Shua,
Lei Jiana,
Bin Xub,
Xueli Zheng*a,
Maolin Yuana,
Haiyan Fua,
Ruixiang Lia and
Hua Chen*a
aKey Lab of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: scuhchen@163.com; zhengxueli@scu.edu.cn
bKey Laboratory of Green Chemistry of Sichuan Institutes of Higher Education, Zigong 643000, China
First published on 31st October 2016
Abstract
Two new phosphorus ligands (L1 and L2) were developed for rhodium-catalyzed hydroformylation of cycloolefins. L1 produced high conversion for cyclohexene (94.7%) and high dialdehyde selectivity for NBD (97.1%) and DCPD (98.4%). Analogue pincer ligands L3–L6 with different steric and electronic character were also investigated in these hydroformylations.
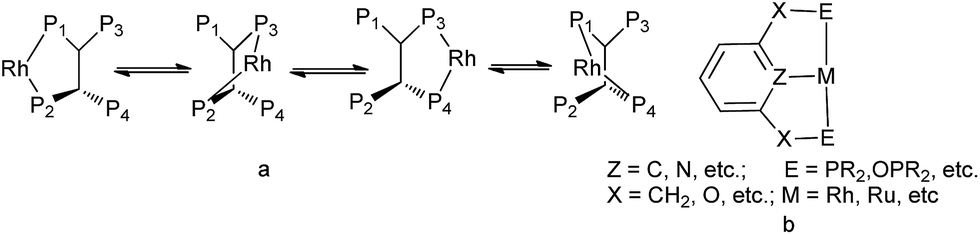
Hydroformylation of olefins is one of the most important homogeneous catalytic reactions and the process is 100% atom-economical.1 Particularly, some special cycloolefins, i.e., cyclohexene, 2,5-norbornadiene (NBD) and dicyclopentadiene (DCPD), have attracted great attention, since their hydroformylation products are of high added value in the manufacture of fine chemicals. Therefore, developing an efficient catalytic system for the above “difficult substrates” is really a challenging work. Usually, the reactivity of a homogenous catalyst increases at the expense of its stability and selectivity, and in addition, the ligand is always a matter of concern. During the past few decades, in order to access high activity and selectivity in rhodium-catalyzed hydroformylation, phosphorous ligands were extensively explored due to its unique stereoelectronic properties, such as bisbi,2 biphephos,3 naphos,4 xantphos,5 and pyrrolyl-substituted multiphosphorus (bis-, tri- and tetra-) ligands.6 Tri- and tetraphosphorus ligands could afford superior regioselectivity to bisanalogue in isomerization–hydroformylation of internal olefins with the presence of Rh(acac)(CO)2.6c,7 The reason was probably that the multi-phosphorus increased the local phosphorus concentration around the metal center and was capable of forming multiple chelating modes with Rh (Fig. 1a), hence their chelating ability was enhanced.6c,7c,8 In comparison, pincer ligands also have good chelating ability with rhodium which preferentially coordinates to transition metal in tridentate chelating mode (Fig. 1b) and serve as excellent precursors in many organic transformations,9 like bond activation,10 dehydrogenation of secondary alcohols to ketones, hydrogenation of esters to alcohols,11 polymerization of olefins12 and etc. Moreover, pincer-based catalyst could often offer high efficiency as well as selectivity.9g However, there are few reports on the application of pincer ligand to rhodium-catalyzed hydroformylation. Different from tri- or tetraphosphorus ligand, pincer ligand could bind tightly to three adjacent coplanar sites of transition metal center9c–f and in addition, its backbone is relatively smaller. It was found that phosphorus ligands possessing strong π-acceptor were effective in terms of regioselectivity and activity in Rh-catalyzed hydroformylation, for example, the pyrrole-based bisphosphoramidites and phosphites could give good performance due to bearing the electron-withdrawing substituent.6,13 In this study, initiated with these pioneer work stated above, PNP pincer ligands L1 and L2 were synthesized and applied to the hydroformylation of cyclohexene, NBD and DCPD. And for comparison, their analogue ligands L3–L6 with different steric and electronic character were also investigated in these hydroformylation.
The pincer ligands L1 and L2 were easily prepared in moderate to good yields via three steps (scheme 1). The diethyl pyridine-2,6-dicarboxylate (B), at first, was easily prepared by adding acetylchloride into a solution of 2,6-pyridinedicarboxylic acid (A) in ethanol at room temperature and stirred for 24 h.14 And then, the reduction of B by NaBH4 afforded the ligand skeleton pyridine-2,6-dimethanol (C).15 Finally, L1 was synthesized through the nucleophilic substitution of chlorodipyrrolylphosphine with (C) under the presence of triethylamine.6b L2 could be synthesized from (C) and phosphorochloridite. Hydroformylation of cycloolefins with the new ligand L1 and L2 were then investigated, respectively. Regardless of double bond isomerization, hydroformylation of cyclohexene only yields cyclohexanecarboxaldehyde, which is a vital intermediate for the production of pharmaceuticals (i.e., melagatran or fosinopril); but, its hydroformylation may encounter harsh reaction conditions or low activity.16 Since the hydroformylation was highly dependent on the reaction conditions, the effects of ligand/metal ratio, temperature and pressure were explored with L1 as the benchmark ligand. The rhodium catalyst was prepared in situ by mixing L1 with Rh(acac)(CO)2 in toluene. And the products of hydroformylation were analyzed by gas chromatography (GC). It should be noted that the hydrogenation product cyclohexane was not detected by GC analysis, and the selectivity to cyclohexanecarboxaldehyde was nearly 100%. As summarized in Table 1, it is not difficult to find that the addition of L1 indeed improved the activity (entries 1–7), which was quite different from the suppressive behavior of SPOs ligand in Rh-catalyzed hydroformylation of cyclo-hexene.16a At lower P/Rh ratios, lower activity was observed (entries 1–4). A minimum ligand/metal ratio of 20 is essential to achieve high conversion. Further increasing the ligand/metal ratio did not improve but reduce the activity (entries 5–7). According to ligand dissociation in Rh-catalyzed hydroformylation,8a increasing the local phosphine concentration around the rhodium atom could enhance the coordination between P and Rh to form complex E (Scheme S1, ESI†), which would benefit the reaction activity. However, more excess ligand would block the coordination site and the coordination of cyclohexene with rhodium active species would become difficult. Therefore, further increasing the ligand/metal ratio would cause its activity to drop. The effects of temperature and CO/H2 pressure were also tested. The reaction rate was accelerated by high temperature (Table 1, entries 4, 8–11). To our delight, the hydroformylation of cyclohexene could be conducted under 80 °C and the conversion could reach up to 83.4% when elongating the reaction time to 5 h (Table 1, entry 9), which was higher than the result reported by Saikia et al. (75.0%, 80 °C, 3.5 MPa, S/C: 500).16b Under the optimized reaction conditions (ligand/metal ratio = 20, 100 °C, CO/H2: 3 MPa), the conversion was around 94.7%. In addition, moderate conversion (68.1–72.6%) could be observed when the S/C increased to 2000 and 3000 (Table 1, entries 14 and 15), and the highest TOF could reach up to 511, which might indicate that the L1/Rh system had a good catalytic activity. Simultaneously, ligand L2 as well as L3–L6 were also investigated in the hydroformylation of cyclohexene with the presence of Rh(acac)(CO)2, and the results under individual optimized P/Rh molar ratio were summarized in Table 2. It can be seen that except for L3 and L4, the other ligands could improve the conversion to approximately 90.2–94.7%. The skeleton of ligands L1, L2 and L3 were similar. The performance of L2/Rh system could be comparable with L1/Rh at the P/Rh molar ratio of 5, which was quite lower than that for L1/Rh system (20), we inferred that this was probably due to the more stereo-hindered structural feature of L2. Fortunately, the plot of cyclohexene conversion vs. P/Rh ratio (see Fig. S2, ESI†) gave some clue that the optimal P/Rh ratio of L2 and L4 was significantly lower than those for L1 and L3, which evidenced that L2 and L4 might be of more steric hindrance. L3/Rh afforded the conversion over 10% lower than L1/Rh, since the electron withdrawing pyrrolyl made the π-acceptor ability of L1 enhanced,9,13 which probably resulted in easier carbon monoxide dissociation from intermediate I to form II and stronger coordination to alkene in intermediate III13a (Scheme S2, ESI†). It is likely that the nitrogen atom in L1–L3 might not coordinate to Rh metal.17 As for L4, the difference is that no oxygen atom connected with the phosphorus atom, which might coordinate with Rh via three sites (P, P, N),9a thus the coordination of L4 to Rh could be stronger than that of L1–L3. Therefore, the dissociation of carbon monoxide and the coordination of cyclohexene in L4/Rh might be more difficult and thus led to lower conversion than L1/Rh and L2/Rh. L5 as well as L6 were likely of medium steric hindrance and afforded good results with medial P/Rh ratio.
 | ||
| Scheme 1 Synthesis of ligands L1 and L2; and the structure of ligands L3–L6. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | P/Rh | T (°C) | P (Mpa) | t (h) | Conv.b (%) | TOFc (h−1) |
| a Reaction condition: S/C = 1000, [Rh] = 2.12 × 10−3 mol L−1, toluene: 2.3 mL, cyclohexene: 0.7 mL.b The conversion of cyclohexene based on GC which was equivalent to the yield of cyclohexanecarboxaldehyde; determined by GC analysis.c The turnover frequency of cyclohexene.d S/C = 2000.e S/C = 3000. | ||||||
| 1 | 0 | 100 | 3 | 3 | 60.0 | 200 |
| 2 | 10 | 100 | 3 | 3 | 75.1 | 250 |
| 3 | 15 | 100 | 3 | 3 | 83.1 | 277 |
| 4 | 20 | 100 | 3 | 3 | 94.7 | 316 |
| 5 | 25 | 100 | 3 | 3 | 93.6 | 312 |
| 6 | 30 | 100 | 3 | 3 | 92.8 | 309 |
| 7 | 35 | 100 | 3 | 3 | 82.1 | 274 |
| 8 | 20 | 80 | 3 | 3 | 38.9 | 130 |
| 9 | 20 | 80 | 3 | 5 | 83.4 | 167 |
| 10 | 20 | 90 | 3 | 3 | 80.1 | 267 |
| 11 | 20 | 110 | 3 | 3 | 96.6 | 322 |
| 12 | 20 | 100 | 2 | 4 | 85.9 | 215 |
| 13 | 20 | 100 | 4 | 2 | 93.9 | 469 |
| 14 | 20 | 100 | 3 | 4 | 72.6d | 363 |
| 15 | 20 | 100 | 3 | 4 | 68.1e | 511 |
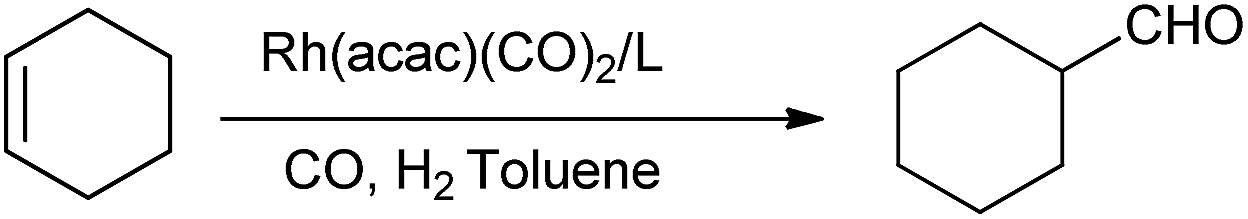
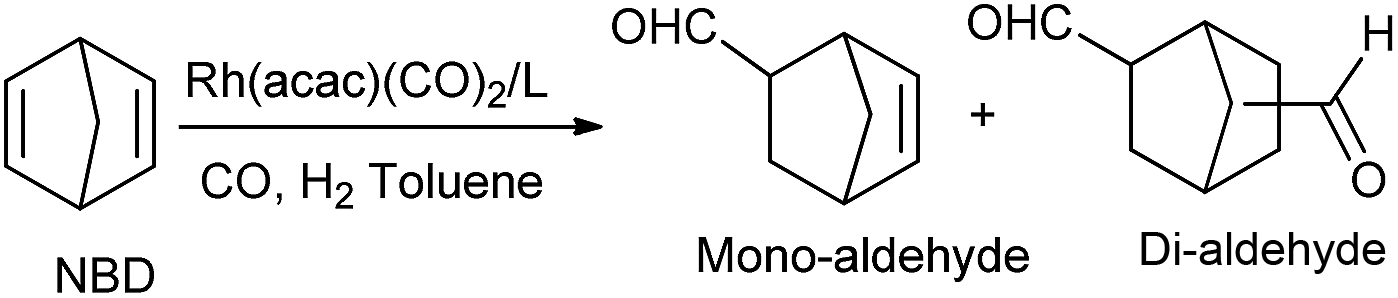
The hydroformylation of 2,5-norbornadiene (NBD) could generate dialdehyde products and its carboxylated derivatives are building blocks for the manufacture of advanced photo-electron instruments.18 On account of its unique stereochemical structure and its ability to form stable complexes with rhodium metal (Scheme S3,† intermediate IX), which unfavors the transformation of the active species VIII,19 as a consequence, its homogeneous hydroformylation usually desired high pressure or prolonged reaction time.20 To our surprise, hydroformylation of NBD with the L/Rh system proceeds much better than that ever reported in homogeneous system.19,20 As shown in Table 3, the behaviour of L1 and L2 was similar to those in cyclohexene. L1 and L2 as well as L6 afforded quite good activity and good selectivity to dialdehyde (around 94.2–97.1%), for instance, when the hydroformylation was conducted at 80 °C with the presence of L2/Rh, nearly 100% of substrate was converted to aldehyde and the selectivity to dialdehyde was up to 96.7% after 3 h (Table 3, entry 3), and at 100 °C the selectivity could reach 96.2% after 1 h, which was superior to the homogeneous catalytic results reported so far.19,20 The pyrrolyl-substitute might improve the π-acceptor ability of L1 and readily caused the dissociation of CO to form VII, thus then the active species VIII was easily formed. Compared to L1, the more bulky L2 would inhibit the formation of intermediate IX. In that case the formation of less stereo-hindered species VIII was favored. Nearly no dialdehyde or low selectivity to aldehyde (0–4.3%) was observed in L3–L5 based system. The inferior π-acceptor ability of L3 (vs. L1) and less steric hindrance (vs. L2), as well as the three coordination sites (P, P, N) of L4 might facilitate the formation of intermediate IX. Intriguingly, the selectivity to mono- or dialdehyde could be reversed by increasing the temperature and prolonging the reaction time in L6/Rh system (Table 3, entries 8–10).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | L | t (h) | T (°C) | Conv.b (%) | Selectivityc (%) | Hydro.d (%) | |
| Mono- | Di- | ||||||
| a S/C = 1000, [Rh] = 2.12 × 10−3 mol L−1, toluene: 2.3 mL, NBD: 0.7 mL, 2 Mpa.b The conversion of NBD based GC.c Selectivity for mono- or di-aldehydes.d Selectivity for hydrogenation products. | |||||||
| 1 | L1 | 3 | 80 | 99.9 | 3.6 | 95.4 | 1.0 |
| 2 | L1 | 1 | 100 | 97.9 | 0.6 | 97.1 | 2.3 |
| 3 | L2 | 3 | 80 | 99.9 | 1.8 | 96.7 | 1.5 |
| 4 | L2 | 1 | 100 | 99.9 | 2.3 | 96.2 | 1.5 |
| 5 | L3 | 4 | 100 | 88.6 | 37.8 | 4.3 | 57.9 |
| 6 | L4 | 6 | 100 | 20.5 | 97.6 | — | 2.4 |
| 7 | L5 | 5 | 100 | 89.5 | 30.2 | — | 69.8 |
| 8 | L6 | 3 | 80 | 25.5 | 98.0 | — | 2.0 |
| 9 | L6 | 1 | 100 | 92.5 | 26.7 | 70.8 | 2.5 |
| 10 | L6 | 3 | 100 | 99.9 | — | 94.2 | 5.8 |
The hydroformylation of dicyclopentadiene (DCPD) could generate tricyclodecanemonoaldehyde (TCDMA) and tricyclodecanedialdehyde (TCDDA), and the potential application of TCDDA is larger than that of TCDMA.21 The hydroformylation might firstly occur on the 8, 9 double bond to generate TCDMA, and subsequently take place at the cyclopentenyl moiety of TCDMA to form TCDDA. As the cyclopentenyl part is less reactive than the norbornenyl moiety,22 the second step is always the bottleneck and suffers from either harsh reaction conditions (22 MPa, 130 °C)23 or adding additives (i.e., a cationic surfactant, a buffer solution) or the necessity of bimetallic catalyst.22a,24 Our previous work found that high conversion (99.9%) and good selectivity (95.0%) to TCDDA could be achieved under 5 MPa and 100 °C in the presence of Rh complex and mixed mono- and bidentate phosphorus ligands.25 The hydroformylation of DCPD using ligands L1–L6 with the presence of Rh(acac)(CO)2 were demonstrated in Table 4. It can be seen that all the ligands exerted good activity and moderate to high selectivity to TCDDA (under heating at 100 °C for 9 h). Fortunately, the selectivity to TCDDA could reach up to 98.4% when L1 was used as the ligand, which is the highest selectivity to TCDDA under the similar reaction conditions ever reported before.25 Intriguingly, the selectivity to mono- or dialdehyde could be abruptly rolled over contingent on the pressure. The selectivity of TCDMA was 99.0% and TCDDA was not detected when conducting the hydroformylation under 80 °C and 1 MPa (shown in Table S1,† entry 1). Apparently, L1/Rh system not only allows highly selective formation of TCDMA or TCDDA under mild conditions, but also avoid the necessity of adding additives or adding mixed mono- and bidentate phosphorus ligands.25 When L2–L4 was used as the ligand, respectively, moderate selectivity to TCDDA was observed (70.5–76.6%), which was probably due to the bulky steric hindrance of L2 and L4, and relatively weaker π-acceptor ability of L3 as mentioned above. In contrast, L5 and L6 were of medium steric hindrance, as a result, their rhodium complex might be less crowded, thereby good selectivity was obtained by using L5 (89.1%) and L6 (92.9%).
|
|||||
|---|---|---|---|---|---|
| Entry | L | Conv.b (%) | Selectivityc | Hydro.d (%) | |
| TCDMA | TCDDA | ||||
| a Conditions: S/C = 1000, [Rh] = 2.12 × 10−3 mol L−1, toluene: 3 mL, 5 Mpa, 100 °C, 9 h.b The conversion of DCPD based on GC.c Selectivity for TCDMA or TCDDA.d See Table 3. | |||||
| 1 | L1 | 99.9 | 0.2 | 98.4 | 1.4 |
| 2 | L2 | 99.9 | 26.6 | 71.1 | 2.3 |
| 3 | L3 | 99.9 | 20.5 | 76.6 | 2.9 |
| 4 | L4 | 99.9 | 23.4 | 70.5 | 6.1 |
| 5 | L5 | 99.9 | 7.6 | 89.1 | 3.3 |
| 6 | L6 | 99.9 | 3.5 | 92.9 | 3.6 |

In summary, as for the difficult substrates like cyclohexene, NBD and DCPD, the demand for a simultaneous increase in (mono- or di-)aldehyde selectivity and activity can be satisfied by the L1/Rh system. The performance of L2/Rh system in cyclohexene and NBD could be comparable with L1/Rh at lower P/Rh molar ratio (5 vs. 20), probably attributed to the more stereo-hindered structural feature, which might also be the reason that L2/Rh system exhibited relatively lower selectivity to TCDDA in the hydroformylation of bulky DCPD. Owing to the weaker π-acceptor ability of L3 (vs. L1) and less steric hindrance (vs. L2), as well as the three coordination sites (P, P, N) of L4, L2 and L4 gave inferior performance in the hydroformylation of these substrates. In comparison, L5 and L6 were likely of medium steric hindrance and afforded good results with medial P/Rh ratio, except for lower selectivity to dialdehyde in NBD made by L5. These results enable the L1/Rh system to be a potential candidate for the hydroformylation of cycloolefins.
Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (No. 201202108), from the Opening Project of Key Laboratory of Green Chemistry of Sichuan Institutes of Higher Education (No. LZJ1402), and from the Sichuan university outstanding scholar research fund (No. 2015SCU04A05).Notes and references
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed.
- (a) W. A. Herrmann, C. W. Kohlpaintner, E. Herdtweck and P. Kiprof, Inorg. Chem., 1991, 30, 427 CrossRef; (b) C. P. Casey, G. T. Whiteker, M. G. Melville, L. M. Petrovich, J. A. Gavney and D. R. Powell, J. Am. Chem. Soc., 1992, 114, 5535 CrossRef CAS; (c) C. P. Casey, E. L. Paulsen, E. W. Beuttenmueller, B. R. Pro, L. M. Petrovich, B. A. Matter and D. R. Powell, J. Am. Chem. Soc., 1997, 119, 11817 CrossRef CAS.
- G. D. Cunny and S. L. Buchwald, J. Am. Chem. Soc., 1993, 115, 2066 CrossRef.
- (a) W. A. Herrmann, R. Schmid, C. W. Kohlpaintner and T. Priermeier, Organometallics, 1995, 14, 1961 CrossRef CAS; (b) H. Klein, R. Jackstell, K.-D. Wiese, C. Borgmann and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 3408 CrossRef CAS.
- (a) L. A. Van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616 CrossRef CAS; (b) L. A. vander Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 336 CrossRef CAS; (c) L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1999, 18, 4765 CrossRef CAS; (d) J. J. Carbó, F. Maseras, C. Bo and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 7630 CrossRef; (e) P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895 CrossRef CAS PubMed.
- (a) R. Jackstell, H. Klein, M. Beller, K.-D. Wiese and D. Rottger, Eur. J. Org. Chem., 2001, 20, 3871 CrossRef; (b) S. C. van der Slot, J. Duran, J. Luten, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 2002, 21, 3873 CrossRef CAS; (c) Y. Yan, X. Zhang and X. M. Zhang, J. Am. Chem. Soc., 2006, 128, 16058 CrossRef CAS PubMed.
- (a) Y. Yan, X. Zhang and X. Zhang, Adv. Synth. Catal., 2007, 349, 1582 CrossRef CAS; (b) S. Yu, Y. Chie and X. Zhang, Adv. Synth. Catal., 2009, 351, 537 CrossRef CAS; (c) S. Yu, X. Zhang, Y. Yan, C. Cai, L. Dai and X. Zhang, Chem.–Eur. J., 2010, 16, 4938 CrossRef CAS PubMed; (d) S. Yu, Y. Chie, Z. Guan and X. Zhang, Org. Lett., 2008, 10, 3469 CrossRef CAS PubMed.
- (a) C. Chen, Y. Qiao, H. Geng and X. Zhang, Org. Lett., 2013, 15, 1048 CrossRef CAS PubMed; (b) C. Chen, P. Li, Z. Hu, H. Wang, H. Zhu, X. Hu, Y. Wang, H. Lv and X. Zhang, Org. Chem. Front., 2014, 1, 947 RSC.
- (a) M. Feller, Y. Diskin-Posner, L. J. W. Shimon, E. Ben-Ari and D. Milstein, Organometallics, 2012, 31, 4083 CrossRef CAS; (b) B. Vabre, M. L. Lambert, A. Petit, D. H. Ess and D. Zargarian, Organometallics, 2012, 31, 6041 CrossRef CAS; (c) W. C. Shih and O. V. Ozerov, Organometallics, 2015, 34, 459 CrossRef; (d) A. J. Kosanorich, J. H. Reibenspies and O. V. Ozerov, Organometallics, 2016, 35, 513 CrossRef; (e) S. D. Timpa, C. J. Pell and O. V. Ozerov, J. Am. Chem. Soc., 2014, 136, 14772 CrossRef CAS PubMed; (f) H. A. Younus, N. Ahmad, W. Su and F. Verpoort, Coord. Chem. Rev., 2014, 276, 112 CrossRef CAS; (g) H. A. Younus, W. Su, N. Ahmad, S. Chen and F. Verpoort, Adv. Synth. Catal., 2015, 357, 283 CrossRef CAS.
- (a) R. Cohen, M. E. van der Boom, L. J. W. Shimon, H. Ro-zenberg and D. Milstein, J. Am. Chem. Soc., 2000, 122, 7723 CrossRef CAS; (b) M. E. Van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (c) M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004 CrossRef CAS PubMed.
- J. Zhang, E. Balaraman, G. Leitus and D. Milstein, Organometallics, 2011, 30, 5716 CrossRef CAS.
- (a) J. Yao, W. T. Wong and G. Jia, J. Organomet. Chem., 2000, 598, 228 CrossRef CAS; (b) A. Alzamly, S. Gambarotta and I. Korobkov, Organometallics, 2013, 32, 7204 CrossRef CAS; (c) S. Sarkar, K. P. McGowan, S. Kuppuswamy, I. Ghiviriga, K. A. Abboud and A. S. Veige, J. Am. Chem. Soc., 2012, 134, 4509 CrossRef CAS PubMed; (d) L. Luconi, J. Klosin, A. J. Smith, S. Germain, E. Schulz, J. Hannedouche and G. Giam-bastiani, Dalton Trans., 2013, 42, 16056 RSC; (e) L. Chen, P. Ai, J. Gu, S. Jie and B. G. Li, J. Organomet. Chem., 2012, 716, 55 CrossRef CAS.
- (a) K. G. Moloy and J. L. Petersen, J. Am. Chem. Soc., 1995, 117, 7696 CrossRef CAS; (b) M. Trzeciak, T. Głowiak, R. Grzybek and J. J. Ziółkowski, J. Chem. Soc., Dalton Trans., 1997, 11, 1831 RSC; (c) O. Diebolt, H. Tricas, Z. Freixa and P. W. N. M. van Leeuwen, ACS Catal., 2013, 3, 128 CrossRef CAS; (d) X. Jia, Z. Wang, C. Xia and K. Ding, Catal. Sci. Technol., 2013, 3, 1901 RSC.
- A. W. Singer and S. M. Mcelvain, J. Am. Chem. Soc., 1935, 57, 1135 CrossRef CAS.
- O. K. Rasheed, P. D. Bailey, A. Lawrence, P. Quayle and J. Raftery, Eur. J. Org. Chem., 2015, 32, 6988 CrossRef.
- (a) A. Christiansen, C. Li, M. Garland, D. Selent, R. Ludwig, R. Franke and A. Börner, ChemCatChem, 2010, 2, 1278 CrossRef CAS; (b) K. Saikia, B. Deb and D. K. Dutta, J. Mol. Catal. A: Chem., 2014, 381, 188 CrossRef CAS; (c) X. Hu, Y. Shi, Y. Zhang, B. Zhu, S. Zhang and W. Huang, Catal. Commun., 2015, 59, 45 CrossRef CAS.
- (a) A. F. Peixoto, A. R. Abreu, A. R. Almeida, A. Neves, P. E. Abreu, M. J. S. M. Moreno, J. C. Bayón, A. A. C. C. Pais and M. M. Pereira, Curr. Org. Synth., 2014, 11, 301 CrossRef CAS; (b) L. Barloy, G. Malaise, S. Ramdeehul, C. Newton, J. A. Osborn and N. Kyritsakas, Inorg. Chem., 2003, 42, 2902 CrossRef CAS PubMed.
- H. Chemical, C. N. Pat. 103,459,365A, 2013.
- X. Yang, H. Liang, H. Fu, X. Zheng, M. Yuang, R. Li and H. Chen, Appl. Organomet. Chem., 2016, 30, 335 CrossRef CAS.
- (a) A. Saus, T. NhuPhu, M. J. Mirbach and M. F. Mirbach, J. Mol. Catal. A: Chem., 1983, 18, 117 CrossRef CAS; (b) C. Botteghi, S. Paganelli, A. Perosa, R. Lazzaroni and G. Uccello-Barretta, J. Organomet. Chem., 1993, 447, 153 CrossRef CAS.
- B. Cornils and B. Payer, Chem.-Ztg., 1974, 98, 70 CAS.
- (a) L. Garlaschelli, M. Marchionna, M. C. Iapalucci and G. Longoni, J. Mol. Catal. A: Chem., 1991, 68, 7 CrossRef CAS; (b) A. M. Trzeciak and J. J. Ziólkowski, J. Organomet. Chem., 1994, 479, 213 CrossRef CAS.
- R. Papp, R. Paciello and C. Benisch, C. N. Pat., 1890203A, 2007.
- (a) X. Pi, Y. Zhou, L. Zhou, M. Yuan, R. Li, H. Fu and H. Chen, Chin. J. Catal., 2011, 32, 566 CrossRef CAS; (b) Y. Fan, S. Ren, B. Xu, T. Shi and Y. Chen, C. N. Pat., 101,053,843 A, 2007.
- R. Luo, H. Liang, X. Zheng, H. Fu, M. Yuan, R. Li and H. Chen, Catal. Commun., 2014, 50, 29 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Ligand synthesis and characterization, and hydroformylation procedure. See DOI: 10.1039/c6ra24144a |
| This journal is © The Royal Society of Chemistry 2016 |