
The electrochemical 4-chlorophenol sensing properties of a plasma-treated multilayer graphene modified photolithography patterned platinum electrode†
Padmanathan Karthick Kannan*a,
Rogerio V. Gelamo b,
Hywel Morganc,
Palaniswamy Sureshd and
Chandra Sekhar Rout*e
b,
Hywel Morganc,
Palaniswamy Sureshd and
Chandra Sekhar Rout*e
aSchool of Chemical Engineering, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: pkk.matsci@gmail.com
bInstituto de Ciências Tecnológicas e Exatas, UFTM, Uberaba, Minas Gerais 38064-200, Brazil
cElectronics and Computer Science, University of Southampton, Southampton SO17 1BJ, UK
dDepartment of Natural Products Chemistry, School of Chemistry, Madurai Kamaraj University, Madurai 625021, Tamil Nadu, India
eSchool of Basic Sciences, Indian Institute of Technology Bhubaneswar, Bhubaneswar, 751013, Odisha, India. E-mail: csrout@iitbbs.ac.in
First published on 31st October 2016
Abstract
The present work describes the electrochemical 4-chlorophenol (4-CP) sensing properties of multilayer graphene samples (MLG). In order to enhance the presence of oxygen functional groups and to increase the edge plane defects, oxygen plasma treatment is adopted. Raman, X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM) and atomic force microscopy (AFM) characterizations are used to investigate the effect of plasma treatment on MLG. Compared to pristine MLG, the oxygen plasma treated one shows increased oxidation current towards 4-CP. The sensor data exhibit high sensitivity and stability with high anti-interference property in the presence of other phenolic compounds including phenol, bisphenol, 2,3-chlorophenol, 2,3,4-nitrophenol and inorganic ions.
Introduction
Chlorophenol isomers such as 2-chlorophenol, 3-chlorophenol and 4-chlorophenol are used widely as fungicides, herbicides, dyes, pharmaceuticals and so on. These chemicals are categorised as environmental pollutants because of their highly toxic nature to aquatic organisms and humans even at very low concentrations.1 Among these chlorinated phenols, 4-chlorophenol (4-CP) has been widely used as a preservative for wood, paints, leathers, dyes and as an intermediate compound for the synthesis of different pharmaceuticals and dyes. However, its release into the environment causes major health problems and contamination of ecosystems. Hence, it is highly desirable to develop a sensor for the rapid detection of 4-CP with high sensitivity and selectivity.For sensing 4-CP, a plethora of techniques including gas chromatography,2 high performance liquid chromatography,3 spectrophotometry4 have been employed. Although, these methods show good sensing performance with high sensitivity and selectivity, there are shortcomings like cumbersome sample preparation or a bulky instrument that makes them unsuitable for the rapid detection of 4-CP in the environment. It is also important to search for an alternative technique for sensing 4-CP. In order to solve these difficulties, electrochemical sensors are highly attractive because of their low cost, easy fabrication, ability to detect analyte molecules with high sensitivity, selectivity and fast response time.5,6 Various nanostructured materials have been investigated for the development of electrochemical sensors.7,8 However, the use of carbon nanomaterials in electrochemical sensors has received great attention because of their cost-effectiveness, wide potential window, lower over potential and high electrocatalytic activity towards various redox molecules.9,10 In addition, these materials also possess high surface to volume ratio, enhanced chemical stability, high electrical conductivity, and biocompatibility with high mechanical strength. Various carbon nanomaterials like carbon nanotubes,11,12 graphene13 and their related nanocomposites14 have been used as sensors. Among these materials, graphene exhibits promising features for the development of electrochemical sensors due to its unique physical and chemical properties. In general, graphene normally exists in two structural regions viz. basal and edge plane. In the basal plane, sp2 conjugated carbon atom are arranged two dimensionally, while edge plane has defective graphitic line of carbon atoms with dangling bonds and various functional moieties like hydroxy, epoxy, carbonyl and carboxyl groups.15 The presence of dangling bonds is responsible for the interaction of graphene with adsorbed chemical species. It has been reported that graphene samples with edge plane shows 7 orders higher electron transfer rate constant than that of basal plane.15 Thus it is very important to tailor the surface of graphene by incorporating functional groups and create large numbers of edge defects. Generally, different methods have been adapted for the functionalization of graphene such as covalent and non-covalent interaction of graphene with organic and inorganic species16 and doping by nitrogen or boron atoms.17,18 Recently, plasma treatment is emerging as a cost effective and facile process for the functionalization of graphene sheets.19,20 The plasma treatment creates functional groups on the graphene surface which internally change the surface reactivity and the electronic structure, leading to the enhanced adsorption of analyte molecules on the surface. It has been reported that plasma treatment effectively reduces the contact resistance at graphene–metal interfaces.21 In addition, plasma treatment creates large number of controlled edge defects and dangling bonds which in turn enhance the rate of the electron transfer reaction or carrier transmission that normally occurs at the electrode–electrolyte interface.
Compared to single layer graphene, multilayer graphene (MLG) possess low electrical resistance, good optical transmittance and high mechanical resistance. In order to generate large numbers of functional groups and defects, MLG samples were treated with oxygen plasma, hence becoming more suitable for electrochemical sensing. To the best of our knowledge, plasma functionalized graphene has not been investigated so far in electrochemical sensing application. This paper focuses on the study of the electrochemical 4-chlorophenol sensing properties of multilayer graphene (MLG) samples. Scheme 1 is a pictorial representation of the preparation of oxygen plasma treated multilayer graphene followed by modification on a photolithographically patterned Pt electrode for the detection of 4-chlorophenol.
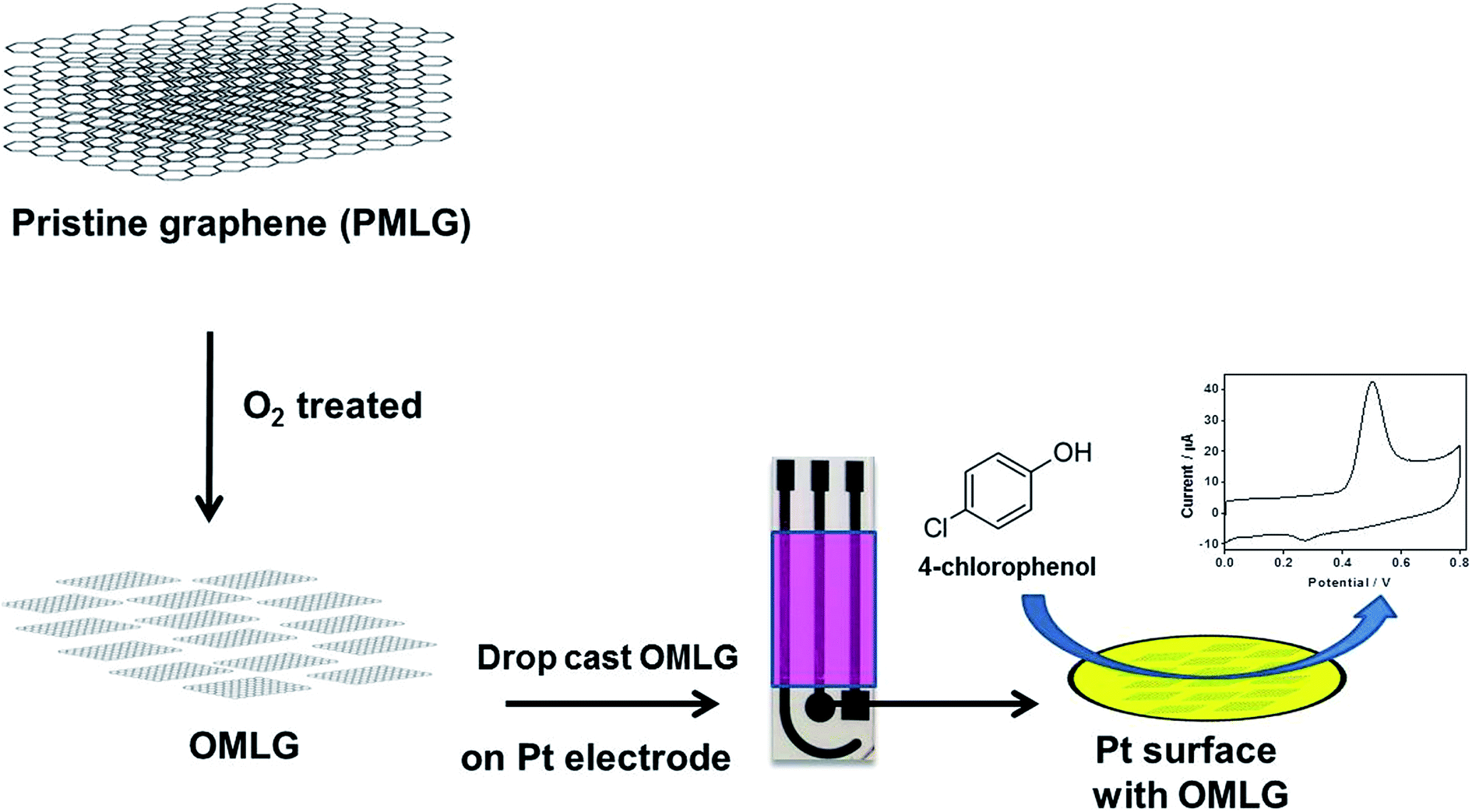 | ||
| Scheme 1 Schematic representation of the developed O2 treated MLG based 4-chlorophenol sensor. | ||
Experimental details
Synthesis of multilayer graphene
Natural graphite obtained from Nacional de Grafite Co., Brazil was used for the synthesis of multilayer graphene (MLG). Detailed information on the preparation of oxygen (O2) treated multilayer graphene is reported elsewhere.22 In a typical method, a custom designed microwave assisted system was used for plasma treatment. The system has a rotary pump and oxygen gas was admitted and controlled by a needle valve. The microwave power supply was 1000 W at 2.45 GHz. The pristine and O2 treated MLG samples will henceforth be denoted as PMLG and OMLG respectively.Characterization techniques and electrochemical measurements
Raman spectra were obtained using the Nano-photon Raman-11 system with 532 nm Ar line laser with power of ∼1 mW. The X-ray Photoelectron Spectroscopy (XPS) measurements were made with a VG Microtech Multi-Lab ESCA-3000. XPS experiments were carried out in Ultra High Vacuum (UHV). For SEM characterization, both the MLG (pristine and plasma treated) samples were deposited on an aluminium coated microscope slide by dip-coating. SEM images were obtained using a Dual Beam FIB/SEM (Focused Ion Beam/Scanning Electron Microscopy, Nanolab Nova 200). All images were obtained in the secondary electron mode, using an electron beam of 5 kV and 0.4 nA. AFM images of the samples were taken by using AFM Shimadzu model SPM 9700 in contact mode. The height profile data of the images were measured using the software “Gwyddion”. All electrochemical measurements were performed using a Palmsens 3 potentiostat/galvanostat instrument. All experiments were performed at room temperature with 0.1 M phosphate buffer solution (PBS), pH = 7 as the supporting electrolyte.Preparation of electrochemical electrodes
The electrochemical measurements were carried out using platinum electrodes comprising working, reference and counter, fabricated on a glass substrate using photolithography – see Fig. S1 in ESI.† For the preparation of an in situ Ag/AgCl reference electrode, silver was electrodeposited onto the Pt electrode using chronoamperometry (applied potential = +0.6 V, time = 300 s) by immersing the electrode in silver nitrate solution. The as-prepared silver electrode was then immersed in 0.1 M KCl solution and a constant potential of +0.3 V applied for 500 s. Deposition of the MLG samples were performed by first cleaning the surface of the working electrode with ethanol. MLG were dispersed by sonication in ethanol (2 mg mL−1) for 30 min. Then, 0.5 μL of the MLG dispersion was drop cast onto the Pt and dried at room temperature.Results and discussion
Raman and XPS characterization
Fig. 1 shows Raman spectrum of PMLG and OMLG samples. Both samples show four distinct peaks viz. D band, G band, D′ band and 2D band; their corresponding peak assignments are shown in Table 1. From the table, it can be observed that there is not much significant peak shift in the spectrum of OMLG compared to PMLG. However, the intensity of D band (also known as defect band) increases, which has been attributed to the formation of large numbers of edge defects due to surface disorder caused by the plasma treatment. This observation also indicates the reduction of graphene layers after plasma functionalization.23 Fig. 2 shows a typical XPS spectrum of PMLG and OMLG. As seen in Fig. 2A, the peak at 284.1 eV corresponds to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, typical of sp2 graphitic networks. The peak at 285.5 eV has been attributed to C–OH bond of MLG. Another two peaks at the B.E value of 286.6 eV, 288.4 eV correspond to the presence of hydroxyl (C–O–C) and carboxyl (C–OOH) groups respectively. In addition to these, there is a small satellite graphitic peak at 290.1 eV. Thus, it can be inferred that PMLG shows all the characteristic graphitic peaks similar to that reported in the literature.20 Fig. 2B shows the XPS spectrum of OMLG sample. The spectrum shows all the peaks as observed for pristine sample. However, the intensity of the peaks corresponding to oxygenated functional group increases with respect to graphitic peaks, suggesting the successful incorporation of oxygen molecules on the surface of graphene layer by plasma treatment.24
C bond, typical of sp2 graphitic networks. The peak at 285.5 eV has been attributed to C–OH bond of MLG. Another two peaks at the B.E value of 286.6 eV, 288.4 eV correspond to the presence of hydroxyl (C–O–C) and carboxyl (C–OOH) groups respectively. In addition to these, there is a small satellite graphitic peak at 290.1 eV. Thus, it can be inferred that PMLG shows all the characteristic graphitic peaks similar to that reported in the literature.20 Fig. 2B shows the XPS spectrum of OMLG sample. The spectrum shows all the peaks as observed for pristine sample. However, the intensity of the peaks corresponding to oxygenated functional group increases with respect to graphitic peaks, suggesting the successful incorporation of oxygen molecules on the surface of graphene layer by plasma treatment.24
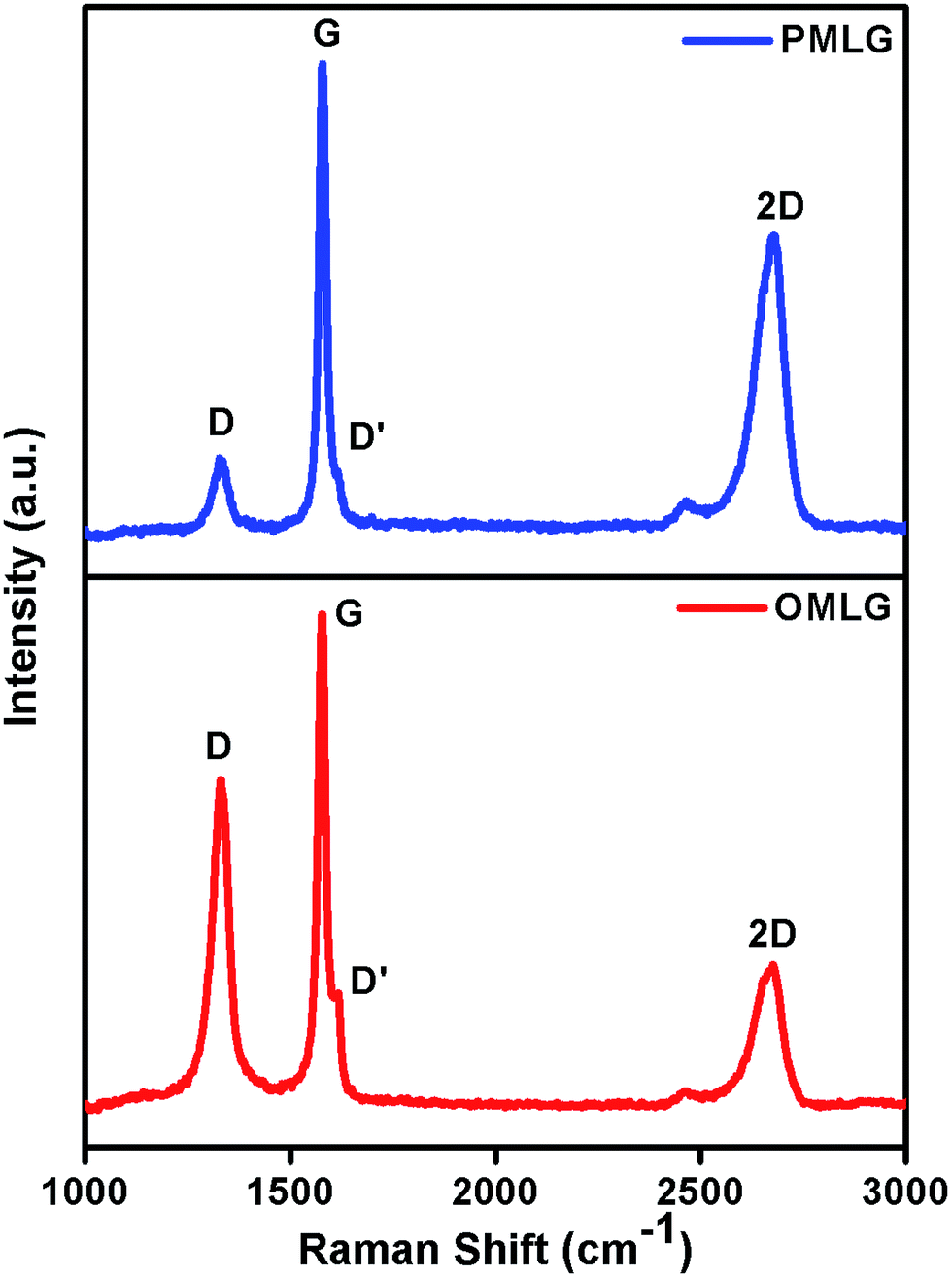 | ||
| Fig. 1 Raman spectrum of PMLG and OMLG samples. | ||
| Sample | Peak position | ID/IG ratio | |||
|---|---|---|---|---|---|
| D | G | D′ | 2D | ||
| PMLG | 1327 cm−1 | 1578 cm−1 | 1607 cm−1 | 2673 cm−1 | 0.42 |
| OMLG | 1330 cm−1 | 1577 cm−1 | 1616 cm−1 | 2667 cm−1 | 0.76 |
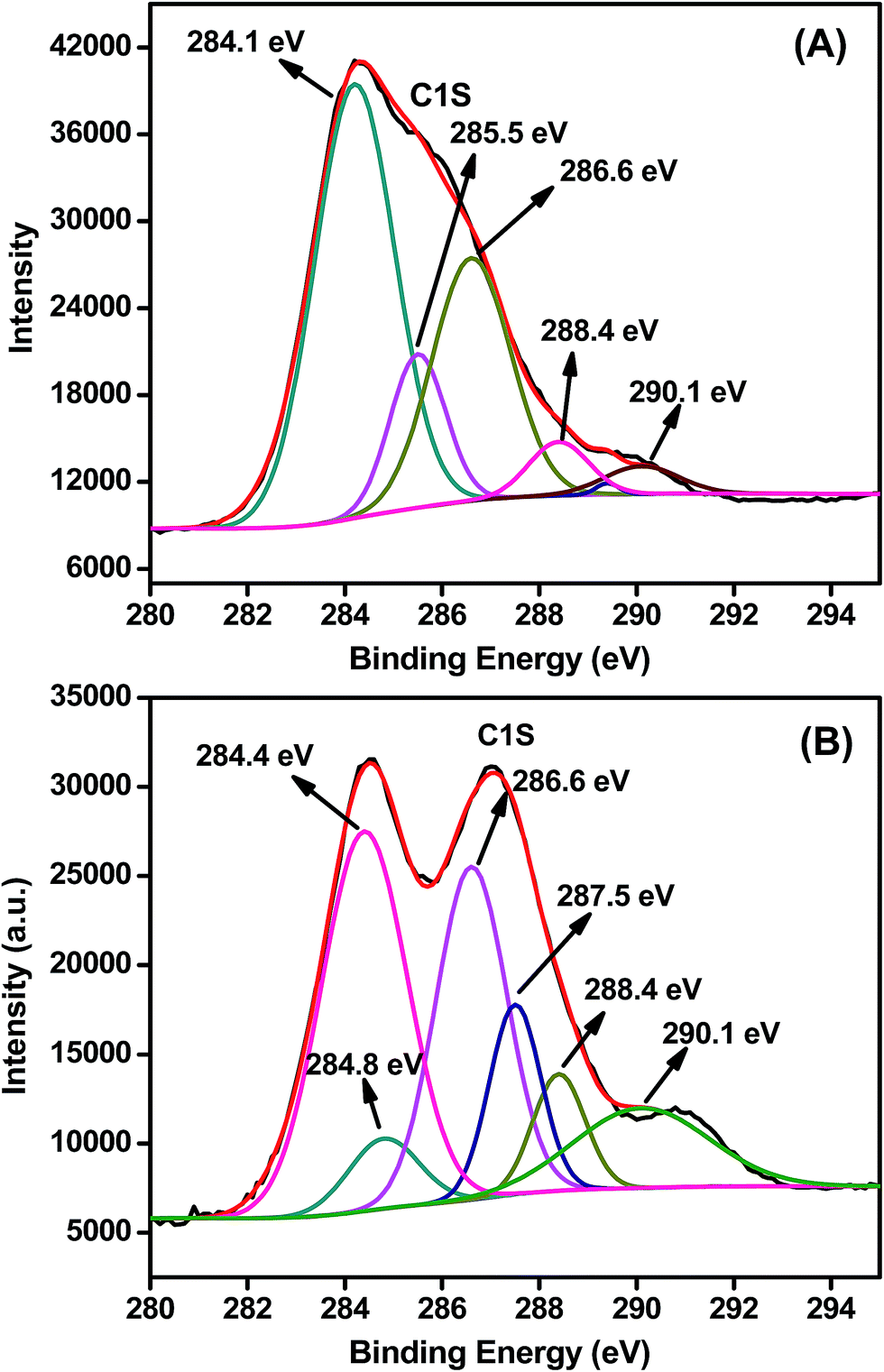 | ||
| Fig. 2 Typical deconvoluted XPS peak of (A) PMLG and (B) OMLG samples. | ||
Morphological characterization
Fig. 3 shows the surface morphology of PMLG and OMLG samples taken with the same magnification. As shown in Fig. 3A, PMLG shows the presence of a thick flake composed of different overlapping graphene layers. On the other hand, OMLG (Fig. 3B) shows uniform distribution of very thin flakes of graphene. These results indicate that due to plasma treatment, graphene samples are highly distorted because of structural disorder.25 As a result, more thin flakes with edge defects are formed compared to PMLG. The presence of active edges on the surface of OMLG can actually enhance the kinetics of the electron transfer reaction occurring on the electrode–electrolyte interface during the electrochemical detection of analyte molecule.26,27 The influence of plasma treatment on the exfoliation of MLG samples was also investigated by AFM. From the images, it can be observed that the graphene flakes in PMLG sample has an approximate thickness of 10 nm, which reduces to 6 nm after oxygen treatment. These results show the occurrence of effective exfoliation due to plasma treatment.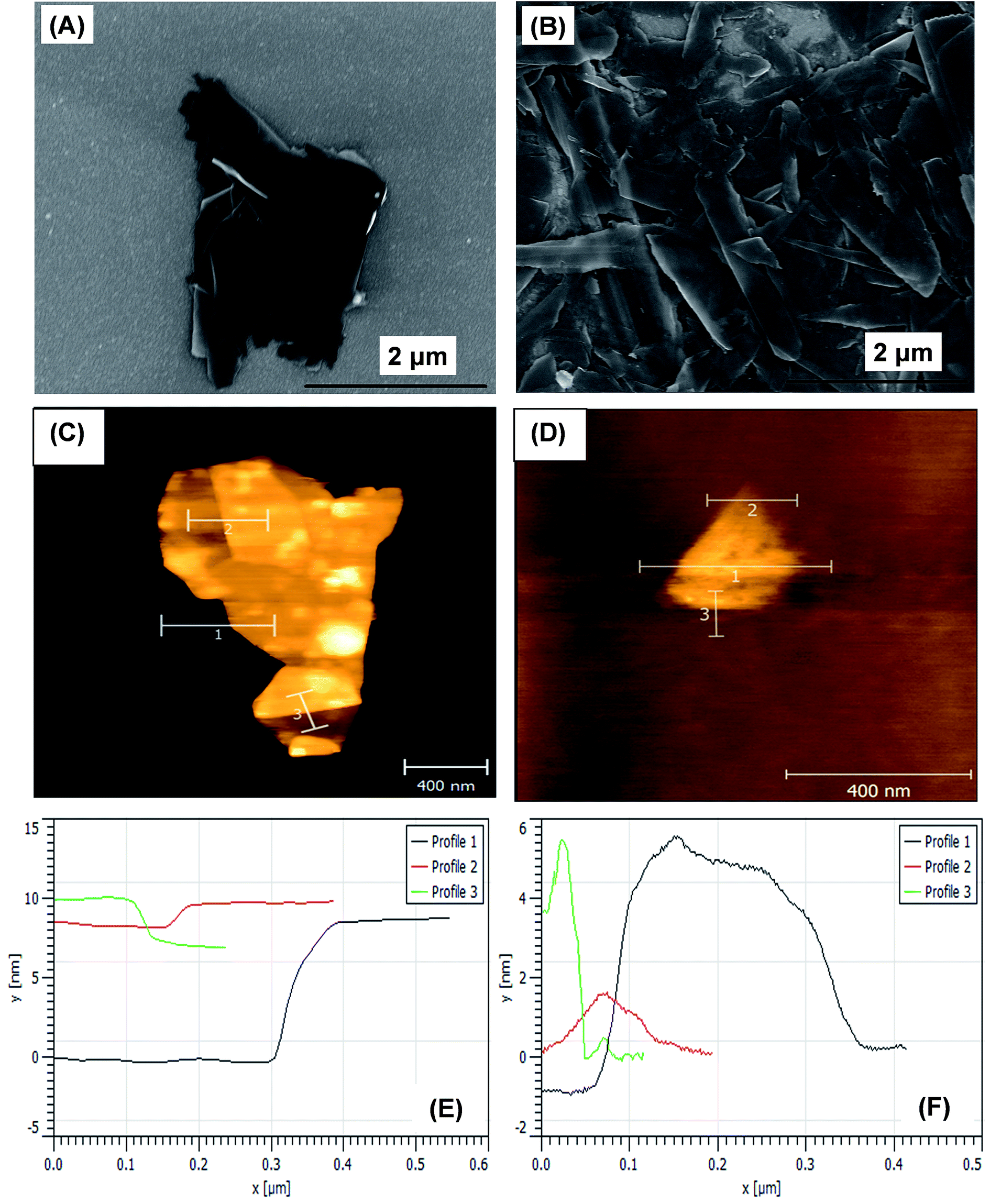 | ||
| Fig. 3 FESEM images of (A) PMLG and (B) OMLG samples. AFM images along with their height profile data (C & E) PMLG and (D & F) OMLG samples. | ||
Electrochemical characterization of MLG samples modified Pt
The electrochemical properties of modified Pt electrodes were measured by performing CV measurements (Fig. 4A) in 0.1 M KCl containing 5 mM of K4[Fe(CN)6] at a scan rate of 50 mV s−1. Bare Pt electrode shows a well-defined redox peak with peak to peak separation voltage (ΔEp) of 110 mV. On the other hand, a PMLG modified electrode shows an enhanced redox peak current compared to bare Pt electrode with ΔEp value of 74 mV indicating the high electrocatalytic activity of PMLG. When the electrode was modified with OMLG, the value of the redox peak current was greatly enhanced with a very low ΔEp value of 65 mV. This observation clearly shows the excellent electrical conductivity and faster electron transfer capability of OMLG compared to other two electrodes. It has been described in a review reported by Dey et al. that it is possible to modify the surface properties of the graphene by plasma treatment method.28 Xie et al. studied the asymmetric surface property of hexane and oxygen treated graphene samples by arranging both the samples on opposite sides. They found that, hexane treated sample shows hydrophobicity while oxygen treated exhibits hydrophilic nature with different electrochemical response.29 Thus, compared to PMLG, OMLG shows enhanced performance because of the surface property improved by the oxygen plasma treatment.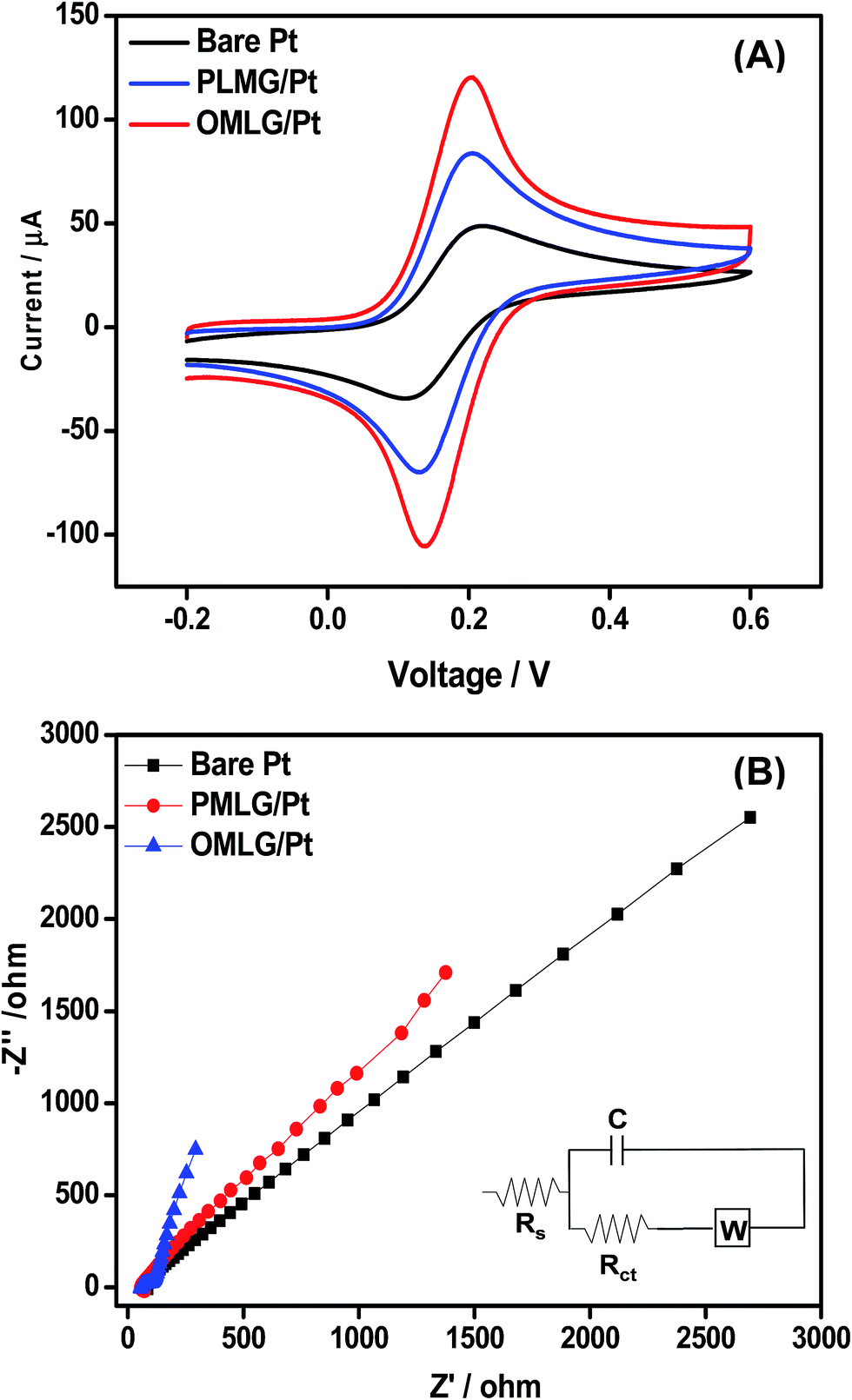 | ||
| Fig. 4 (A) CV data of bare and MLG modified electrodes measured in 0.1 M KCl solution containing 5 mM of K4[Fe(CN)6] at a scan rate of 50 mV s−1. (B) Nyquist plots measured in 0.1 M KCl solution containing 5 mM K4[Fe(CN)6] for bare and MLG modified Pt electrodes. Frequency range: 1 Hz to 50 KHz. Applied AC voltage – 1 mV. Bottom inset is the most fitted equivalent circuit. | ||
The effective surface area of the modified electrodes can be calculated from the CV data of a reversible electrochemical process using the Randles–Sevcik equation 30
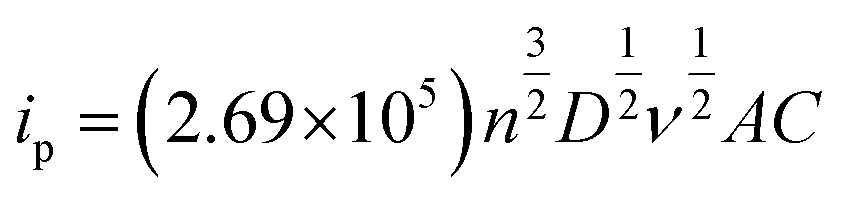 | (1) |
The electrochemical interfacial properties of bare and modified Pt electrodes were ascertained by carrying out electrochemical impedance spectroscopy (EIS) measurements with K3[Fe(CN)6] as a probe. Fig. 4B shows the Nyquist plots of bare and MLG modified Pt electrodes. Bare Pt shows a straight line over the whole region whiles the PMLG and OMLG modified electrodes show a depressed straight line with very low impedance. Compared to PMLG, O2 treated samples have low impedance magnitude, indicating a highly conducting material. The electrical parameters were obtained from the impedance data by fitting to an equivalent circuit shown in bottom inset of Fig. 4B. The circuit consists of the solution resistance (Rs), charge transfer resistance (Rct) with capacitance (C) and Warburg impedance (W). Rct determines the charge transfer ability of a redox probe and the values were found to be 46 Ω, 35 Ω and 10 Ω for bare, PMLG and OMLG modified electrodes respectively. These results show that compared to bare and PMLG modified Pt electrodes, OMLG has a very low Rct which has been ascribed to faster electron transfer capability and large surface to volume ratio. These observations also indicate that OMLG exhibits higher kinetics in the redox process compared to the pristine one. This is due to the fact that during the plasma treatment, the MLG layers are ruptured leading to exfoliation. This leads to the formation of very thin graphene flakes with large number of edge sites and defects which help to accelerate the electrochemical redox reaction.31 The formation of edge defects due to plasma treatment is found to be in agreement with the Raman spectra.
Electrochemical sensing of 4-CP
Fig. 5 shows CV data of bare and MLG modified Pt electrodes measured in 0.1 M PBS (pH 7) containing 0.1 mM 4-CP. Fig. 5A shows that bare Pt does not have any distinct redox peaks in the absence or presence of 4-CP. On the other hand, PMLG and OMLG modified Pt electrodes show a well-defined irreversible oxidation peak (O1) at 0.51 V implying that MLG samples exhibit high electrocatalytic activity in the presence of 4-CP. The oxidation peak potential was shifted towards negative side compared to bare Pt indicating that MLG acts as an effective mediator for the electrocatalytic oxidation of 4-CP. During the second cycle, a small oxidation peak (O2) appeared at 0.27 V, while in the respective negative scan, a small reduction peak (R2) is observed at 0.27 V ascribed to the reversibility of O1 peak.32 Several reports show similar observation and based on their results, electrochemical oxidation reaction occurring at MLG modified Pt electrodes can be described as follows.32,33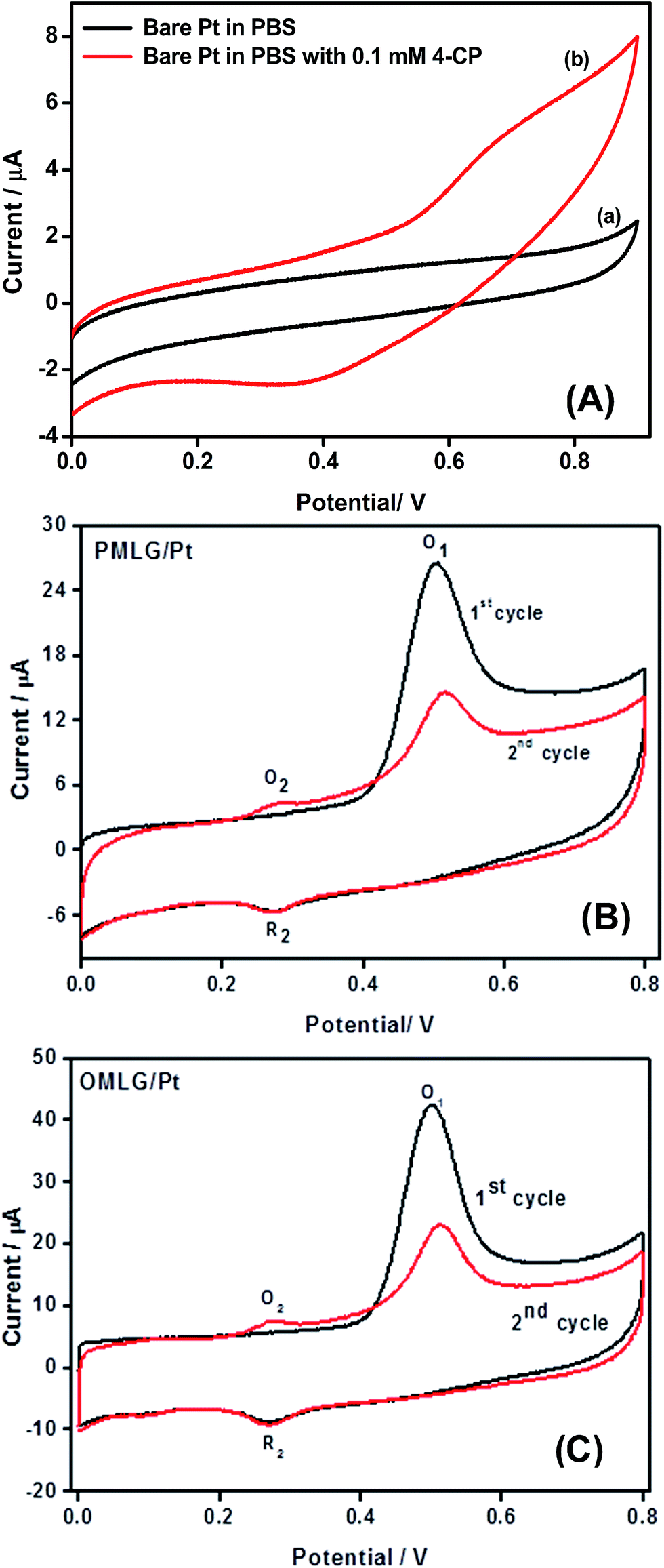 | ||
| Fig. 5 (A) CV data obtained for bare Pt in 0.1 M PBS in the absence (a) and presence (b) of 0.1 mM 4-CP. CV data obtained for (B) PMLG and (C) OMLG samples modified Pt electrode in 0.1 M PBS solution containing 0.1 mM 4-CP. Scan rate – 50 mV s−1. | ||
During the first anodic scan, the phenolic hydroxyl group in 4-CP undergoes oxidation and transformed as phenolic hydroxyl radical and due to this oxidation reaction, peak O1 is appeared.
In the second scanning, anodic peak O2 is occurred due to the oxidation of phenolic hydroxyl radical into benzoquinone. The peak R2 is corresponding to the reversibility of peak O2 which is nothing but the conversion of benzoquinone to hydroquinone by electrochemical reduction reaction. The overall chlorophenol oxidation reaction mechanism can be represented in Scheme 2.
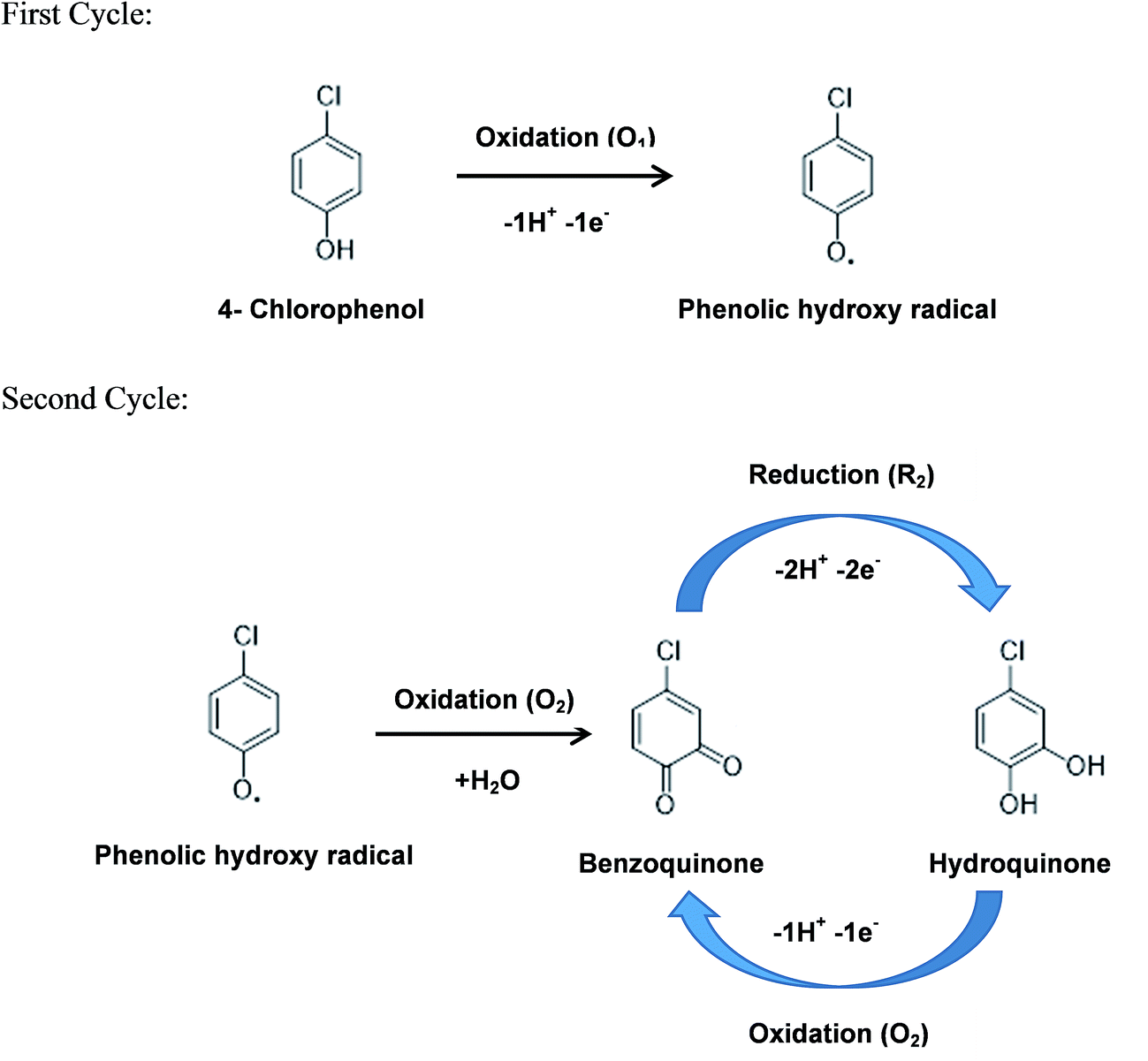 | ||
| Scheme 2 The overall electrochemical reaction mechanism of 4-chlorophenol occurring at OMLG modified Pt electrodes. | ||
Another interesting observation is that after the completion of the first cycle, the current corresponding to O1 peak is decreases, due to fouling of the electrode due to adsorption of products formed by chlorophenol oxidation (phenolic hydroxyl radical) on the surface of Pt electrodes. Compared to PMLG, OMLG shows high oxidation current which can be attributed to the presence of more active edge sites than that of the pristine. Hence, OMLG was chosen for further optimization of the sensor.
Fig. 6A shows the CVs measured for OMLG modified Pt electrode in 0.1 mM 4-CP at different potential scan rates. From the data, the anodic and cathodic peak current can be plotted against the square root of scan rate (Fig. 6B). This is linear over the range of 30 mV s−1 to 500 mV s−1. The corresponding linear regression equations are Ipa = −34.08(ν1/2) + 8.31 and Ipc = 36.09(ν1/2) − 6.53 with a correlation coefficient of R2 = 0.99. These results suggest that the electrochemical oxidation reaction of 4-CP on OMLG modified electrodes is diffusion controlled. Similarly, the change in natural logarithm value of scan rate (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ν) against the anodic peak potential (Epa) of 4-chlorophenol is shown in Fig. 6C. It is observed that, Epa shifts positively with respect to scan rate in the range of 30–500 mV s−1 indicating that the oxidation process of 4-chlorophenol on the electrode surface is an irreversible reaction. The respective linear plot can be expressed as Epa = 0.02615(lnν) + 0.42.
ν) against the anodic peak potential (Epa) of 4-chlorophenol is shown in Fig. 6C. It is observed that, Epa shifts positively with respect to scan rate in the range of 30–500 mV s−1 indicating that the oxidation process of 4-chlorophenol on the electrode surface is an irreversible reaction. The respective linear plot can be expressed as Epa = 0.02615(lnν) + 0.42.
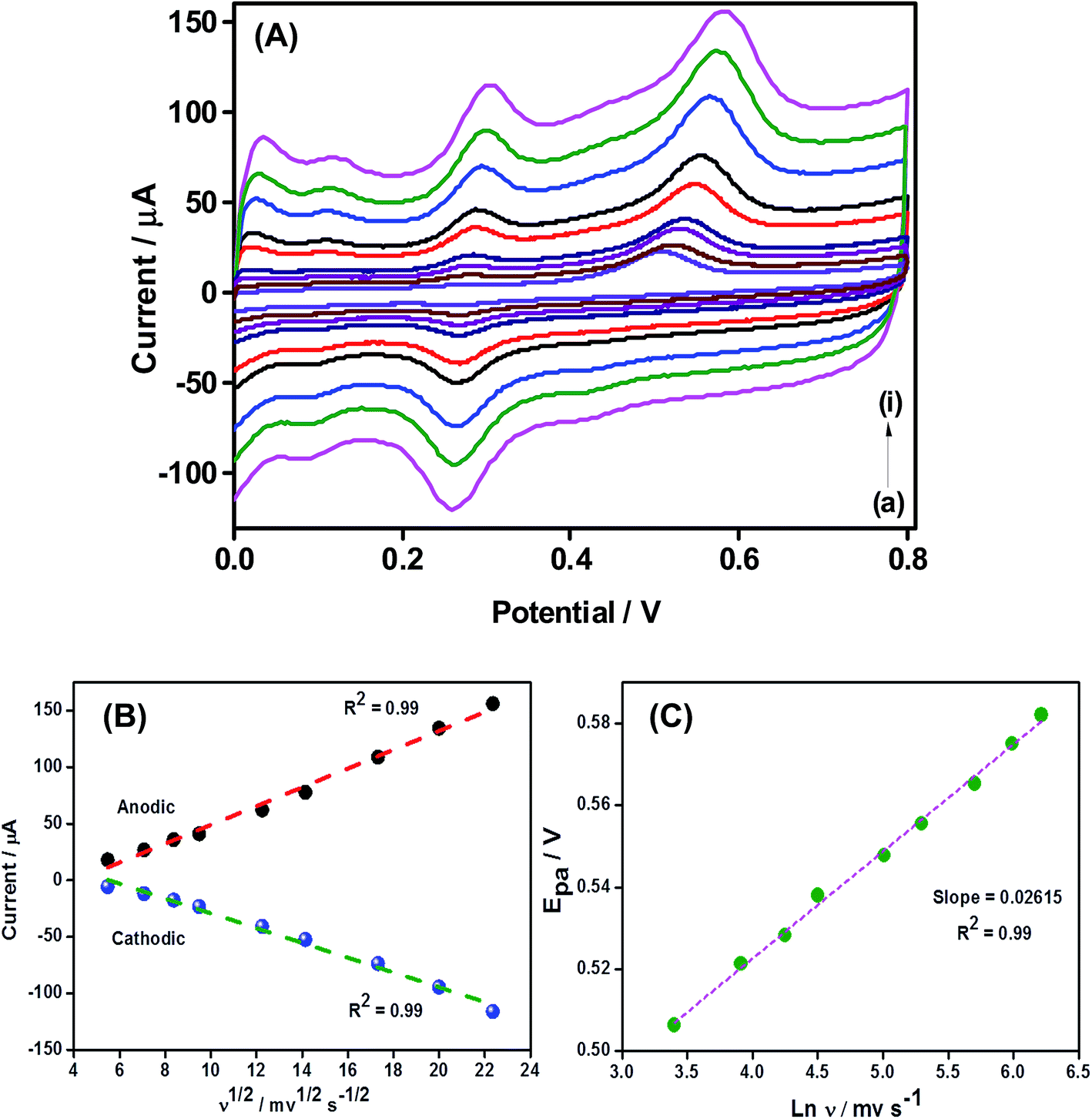 | ||
| Fig. 6 (A) CV data obtained for OMLG modified Pt electrode in 0.1 M PBS containing 0.1 mM 4-CP at different scan-rates in mV s−1: (a) 30, (b) 50, (c) 70, (d) 90, (e) 150, (f) 200, (g) 300, (h) 400, (i) 500. (B) A plot of 4-chlorophenol oxidation and reduction peak current against the square root of the scan-rate. (C) A plot of anodic peak potential value of 4-chlorophenol against the natural logarithm of scan rate. | ||
Fig. 7A shows the CA response of an OMLG/Pt electrode to the successive addition of different concentrations of 4-CP in 0.1 M PBS at an applied voltage of 0.5 V. As shown, the chlorophenol oxidation current increases with increasing concentration. After the addition of 4-CP, the sensor attains equilibrium within 10 s showing a fast response. A calibration plot is shown inset in Fig. 7A with a linear range from 0.5 μM to 10 μM (R2 = 0.99). For higher concentrations, the current gradually increases and at 25 μM, the sensor is saturated which might be due to the adsorption of more 4-CP molecules on the active sites of MLG,34 decreasing the current accordingly. The sensitivity, limit of detection (LOD) and limit of quantification (LOQ) of the sensor were calculated by using the formula as given in ref. 35 and found to be 1.21 μA μM−1 cm−2, 54 nM and 0.18 μM respectively. Table 2 shows the comparison of the performance of other nanomaterials based chlorophenol sensors with the present work. From the table, it can be seen that the developed sensor based on plasma functionalized MLG showed good sensing performance in terms of very low detection limit and detection sensitivity.
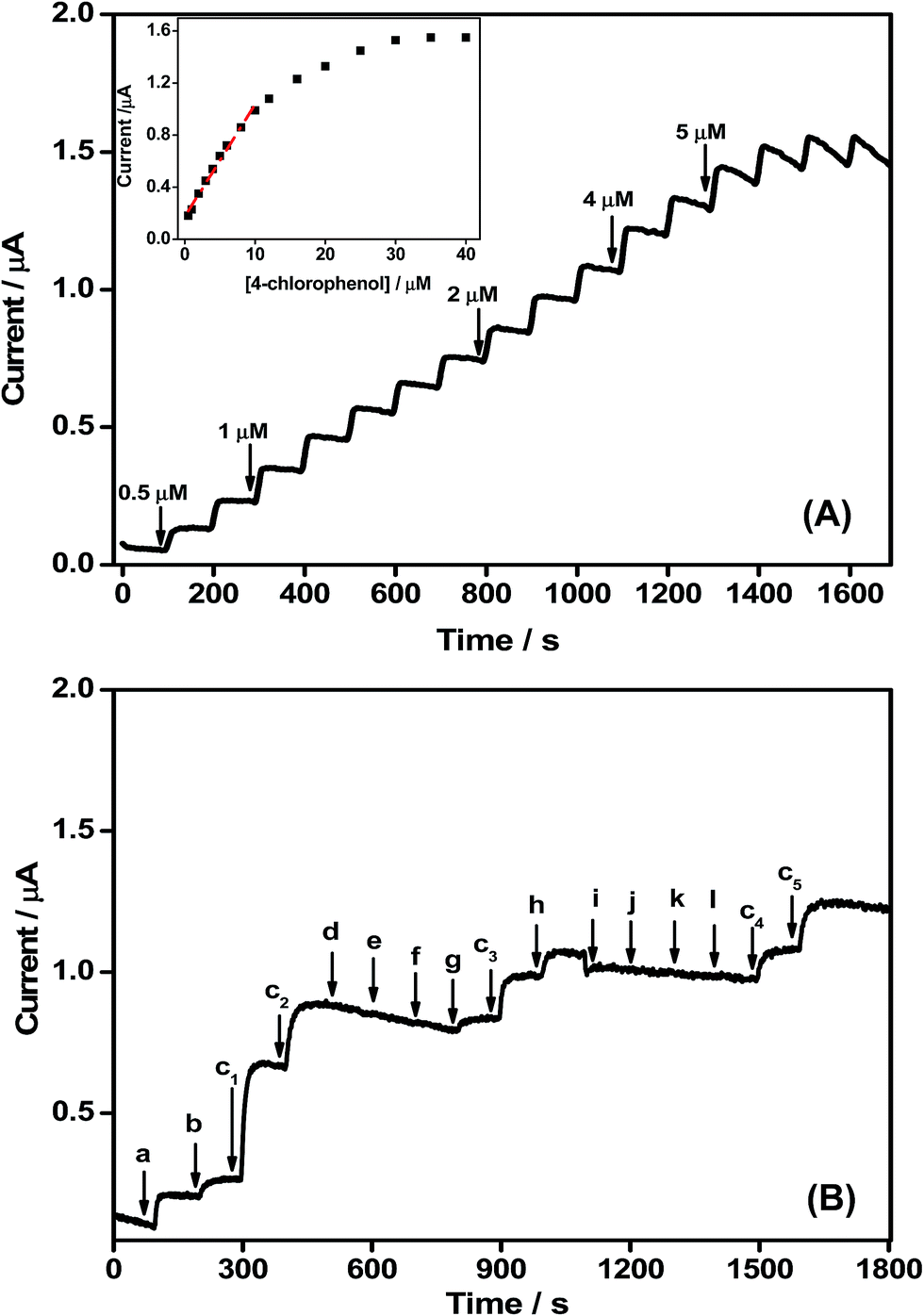 | ||
| Fig. 7 (A) CA curve obtained for OMLG sample modified Pt electrode in 0.1 M PBS (pH 7.0) containing different concentrations of 4-CP. Inset – corresponding calibration data (B) interference data of OMLG sample modified Pt electrode measured in 0.1 M PBS (pH 7) with 0.5 μM of 4-CP (C1–C5) in the presence of 5 μM (a) 3-chlorophenol (b) 2-chlorophenol (d) 2-nitrophenol (e) 3-nitrophenol (f) 4-nitrophenol (g) bisphenol (h) phenol (i) Zn2+ (j) Na+ (k) K+. Applied potential 0.5 V. | ||
| Electrode | Sensitivity (μA μM−1 cm−2) | Calibration technique | Linear range (μM) | LOD (nM) | Ref. |
|---|---|---|---|---|---|
| a DPV – differential pulse voltammetry; CA – chronoamperometry; GCE – glassy carbon electrode; ITO – indium tin oxide electrode; MWCNT – multi-walled carbon tube. | |||||
| Nano-Au/GCE | 0.000397 | CA | 200–4800 | 110 | 36 |
| Horseradish peroxidase/Au/GCE | 0.00396 | CA | 2.5–40 | 390 | 37 |
| 3D ordered macroporous polycysteine/GCE | — | DPV | 0.05–3 | 16.7 | 38 |
| Au/phosphotungstic acid/chitosan/ITO | 0.1247 | DPV | 1–400 | 340 | 39 |
| Au/MWCNT/GCE | — | DPV | 0.3–400 | 110 | 40 |
| MLG/Pt | 1.21 | CA | 0.5–10 | 54 | Present work |
Selectivity is important in assessing the performance of the sensor. Fig. 7B shows the CA response of OMLG/Pt electrode to the successive addition of 0.5 μM 4-CP with 5 μM of interferants at regular intervals of 100 s (in 0.1 M PBS at an applied potential of 0.5 V). The sensor current increases in with the addition of 5 μM of 2-CP, 3-CP and phenol. The current change in the presence of 10 times higher concentrations of 2-CP and 3-CP is much less than the addition of 0.5 μM 4-CP. Moreover, the addition of other interferants such as 2,3,4 nitrophenols, bisphenol and inorganic ions viz. Zn2+, Na+, K+ does not affect the sensor response. Thus the OMLG/Pt sensor has good selectivity towards 4-CP in the presence of potential interfering species. The reproducibility of the OMLG/Pt sensor was evaluated using five similar electrodes prepared under identical conditions in 0.1 M PBS solution containing 0.1 mM 4-CP. An RSD value of 2.3% was obtained, suggesting good reproducibility of the OMLG film. The long term stability of OMLG/Pt was ascertained by measuring the chronoamperometric response over an hour. Fig. S4 in ESI† shows the current for the OMLG/Pt sensor (in 0.1 M PBS with 10 μM 4-CP at an applied potential value of 0.5 V).
The feasibility of the developed sensor in real time analysis is demonstrated by determining the 4-CP sensing performance in tap water using standard addition method. The respective data is shown in Table S1 in ESI.† From the table, it is observed that about 95% recovery is possible with the RSD value of 2% indicating the superior performance of the developed sensor.
Conclusions
An electrochemical sensor for the detection of 4-CP has been developed using multilayer graphene (MLG). In order to enhance the electron transfer property as well as to increase the edge plane defects, MLG samples were treated by oxygen plasma. The increase in the level of edge plane defects as well as functional groups was confirmed by XPS and RAMAN. A standard redox probe was used to study the electron transfer behaviour of pristine and oxygen treated MLG samples. The electrochemical sensing of 4-chlorophenol was studied using oxygen treated MLG modified Pt electrodes. The sensor had high sensitivity, long term stability and good reproducibility with good anti-interference property in the presence of other phenolic compounds.Acknowledgements
Dr PKK sincerely thanks the University Grant commission, India-UK-India Education and Research Initiative (UGC-UKIERI) for the financial assistance to visit University of Southampton, United Kingdom. Dr CSR gratefully acknowledge the University Grant commission, India-UK-India Education and Research Initiative (UGC-UKIERI, Grant No. UGC-2013-14/005) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq/Brazil) for the financial support. Dr PKK also wishes to thank Dr B. Dinesh, NPDF fellow, Department of Chemistry, School of Advanced Sciences, VIT University, Tamil Nadu for several discussions during manuscript preparation.References
- E. O. Igbinosa, E. E. Odjadjare, V. N. Chigor, I. H. Igbinosa, a. O. Emoghene, F. O. Ekhaise, N. O. Igiehon and O. G. Idemudia, Sci. World J., 2013, 2013, 1–11 Search PubMed.
- K.-D. Wang, P.-S. Chen and S.-D. Huang, Anal. Bioanal. Chem., 2013, 406, 2123–2131 CrossRef PubMed.
- R. Alizadeh, Talanta, 2016, 146, 831–838 CrossRef CAS PubMed.
- P. Ncube, R. W. Krause and B. B. Mamba, Sensors, 2011, 11, 4598–4608 CrossRef CAS PubMed.
- C. M. a. Brett, Pure Appl. Chem., 2001, 73, 1969–1977 CrossRef CAS.
- S. Zhang, Z. Shi, J. Wang, Q. Cheng and K. Wu, Electrochim. Acta, 2014, 130, 734–739 CrossRef CAS.
- X.-Y. Yu, Z.-G. Liu and X.-J. Huang, Trends Environ. Anal. Chem., 2014, 3–4, 28–35 CrossRef CAS.
- M. Pumera, Nanomaterials for Electrochemical Sensing and Biosensing, CRC Press, 2014 Search PubMed.
- C. Yang, M. E. Denno, P. Pyakurel and B. J. Venton, Anal. Chim. Acta, 2015, 887, 17–37 CrossRef CAS PubMed.
- M. Zhou and S. Guo, ChemCatChem, 2015, 7, 2744–2764 CrossRef CAS.
- K. Gong, Y. Yan, M. Zhang, L. Su, S. Xiong and L. Mao, Anal. Sci., 2005, 21, 1383–1393 CrossRef CAS PubMed.
- Q. Zhao, Z. Gan and Q. Zhuang, Electroanalysis, 2002, 14, 1609–1613 CrossRef CAS.
- D. Zheng, H. Hu, X. Liu and S. Hu, Curr. Opin. Colloid Interface Sci., 2015, 20, 383–405 CrossRef CAS.
- S. Ge, F. Lan, F. Yu and J. Yu, New J. Chem., 2015, 39, 2380–2395 RSC.
- W. Yuan, Y. Zhou, Y. Li, C. Li, H. Peng, J. Zhang, Z. Liu, L. Dai and G. Shi, Sci. Rep., 2013, 3, 2248 Search PubMed.
- K. S. Subrahmanyam, A. Ghosh, A. Gomathi, A. Govindaraj and C. N. R. Rao, Nanosci. Nanotechnol. Lett., 2009, 1, 28–31 CrossRef CAS.
- Z.-H. Sheng, H.-L. Gao, W.-J. Bao, F.-B. Wang and X.-H. Xia, J. Mater. Chem., 2012, 22, 390–395 RSC.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- A. Nourbakhsh, M. Cantoro, T. Vosch, G. Pourtois, F. Clemente, M. H. van der Veen, J. Hofkens, M. M. Heyns, S. De Gendt and B. F. Sels, Nanotechnology, 2010, 21, 435203 CrossRef PubMed.
- L. Liu, D. Xie, M. Wu, X. Yang, Z. Xu, W. Wang, X. Bai and E. Wang, Carbon, 2012, 50, 3039–3044 CrossRef CAS.
- D. W. Yue, C. H. Ra, X. C. Liu, D. Y. Lee and W. J. Yoo, Nanoscale, 2015, 7, 825–831 RSC.
- R. T. Khare, R. V. Gelamo, M. A. More, D. J. Late and C. S. Rout, Appl. Phys. Lett., 2015, 107, 123503 CrossRef.
- C. Y. Zhi, X. D. Bai and E. G. Wang, Appl. Phys. Lett., 2014, 81, 1690 CrossRef.
- N. Mcevoy, H. Nolan and G. S. Duesberg, Carbon, 2012, 54, 283–290 CrossRef.
- C. Shen, G. Huang, Y. Cheng, R. Cao, F. Ding, U. Schwingenschlögl and Y. Mei, Nanoscale Res. Lett., 2012, 7, 1–8 CrossRef PubMed.
- R. J. Rice and R. L. McCreery, Anal. Chem., 1989, 61, 1637–1641 CrossRef CAS.
- R. R. Moore, C. E. Banks and R. G. Compton, Analyst, 2004, 129, 755–758 RSC.
- A. Dey, A. Chroneos, N. S. J. Braithwaite, R. P. Gandhiraman and S. Krishnamurthy, Appl. Phys. Rev., 2016, 3, 021301 Search PubMed.
- X. Xie, L. Qu, C. Zhou, Y. Li, J. Zhu, H. Bai, G. Shi and L. Dai, ACS Nano, 2010, 4, 6050–6054 CrossRef CAS PubMed.
- P. Zanello, Inorganic electrochemistry: theory, practice and applications, Royal Society of Chemistry, 2003 Search PubMed.
- G. P. Keeley, N. Mcevoy, H. Nolan, M. Holzinger, S. Cosnier and G. S. Duesberg, Chem. Mater., 2014, 26, 1807–1812 CrossRef CAS.
- T. A. Enache and A. M. Oliveira-brett, J. Electroanal. Chem., 2011, 655, 9–16 CrossRef CAS.
- Y. Zhang, Q. Cheng, M. Zheng, X. Liu and K. Wu, J. Hazard. Mater., 2016, 307, 36–42 CrossRef CAS PubMed.
- S. M. Golabi and H. R. Zare, J. Electroanal. Chem., 1999, 465, 168–176 CrossRef CAS.
- P. K. Kannan and C. S. Rout, Chem.–Eur. J., 2015, 21, 9355–9359 CrossRef CAS PubMed.
- C. Qiu, X. Dong, H. Ma and S. Hou, Electroanalysis, 2012, 24, 1201–1206 CrossRef CAS.
- C. Qiu, T. Chen, X. Wang, Y. Li and H. Ma, Colloids Surf., B, 2013, 103, 129–135 CrossRef CAS PubMed.
- S. Zhang, Z. Shi, J. Wang, Q. Cheng and K. Wu, Electrochim. Acta, 2014, 130, 734–739 CrossRef CAS.
- Y. Zhu, M. Wu and S. Liu, J. Appl. Electrochem., 2013, 43, 1035–1041 CrossRef CAS.
- L. Wang, Q. Sun, Y. Liu and Z. Lu, RSC Adv., 2016, 6, 34692–34698 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24136k |
| This journal is © The Royal Society of Chemistry 2016 |