DOI:
10.1039/C6RA24113A
(Paper)
RSC Adv., 2016,
6, 102296-102305
Solvatochromic, thermochromic and pH-sensory DCDHF-hydrazone molecular switch: response to alkaline analytes†
Received
28th September 2016
, Accepted 20th October 2016
First published on 21st October 2016
Abstract
Multi-stimuli responsive DCDHF-hydrazone molecular switch containing a hydrazone recognition moiety is developed for the naked-eye detection of alkaline analytes in both vapour and aqueous media. Mechanisms accounting for the thermochromism, pH-sensitivity and solvatochromism are proposed. DCDHF-hydrazone chromophores of different substituents introduced nanostructures with different morphologies via a re-precipitation technique. The films formulated from these nanostructures function as solid-state vapochromic sensors for probing alkaline vapours such as amines and ammonia. The sensing performance is reversible and has differential sensitivity towards a variety of amines at comparable concentrations. The structure of the hydrazone molecular switch was established spectroscopically and by single crystal X-ray crystallography.
Introduction
Stimuli responsive materials are significant as they can potentially afford smart materials that can be applied in a variety of nanotechnological to pharmacological applications.1 Molecular switches can undergo molecular structure variations in response to external physical or chemical stimuli such as light, pH, and chemical agents.2 The hydrazone-based molecular switches are characterized by their modularity, stability, and simple preparation.3,4 Owing to their structure influenced largely by the type and position of substituent groups, hydrazones can be employed in various fields including organic synthesis,5 medicinal chemistry,6 metal and covalent organic frameworks,7 dynamic combinatorial chemistry,8 dyes and pigments,9 probes,3,10 nonlinear optics,11 and hole-transporting materials in organic light emitting diodes.12 The hydrazone unit structure is correlated to the structure of ketones and aldehydes by replacing the oxygen with the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH– group.3 The hydrazone can be considered for molecular switching due to its inclusion with am imine group that can suffer stimuli responsive cis/trans isomerisation and/or ability to lose proton (Brønsted acid). Hydrazones are usually prepared by three main synthetic pathways: (i) coupling between aryl diazonium salts and β-keto esters or acids active methylene-containing materials, which is also known as the Japp–Klingemann reaction, (ii) condensation of hydrazines and ketones or aldehydes, and (iii) coupling between aryl halides and non-substituted hydrazones.3,4,13–16 On the other hand, DCDHF (2-(dicyanomethylene)-2,5-dihydro-4,5,5-trimethylfuran-3-carbonitrile) chromophores with push-π-pull molecular structure have attracted much attention because of their inherent non-linear optical properties, which are highly sensitive to changes in the environment involving solvent polarity or pH of media, due to their intrinsic character. They have been intensively developed as photo- and electroluminescent materials in many fields including electro-optics, photorefractives, single molecule fluorophores, dye lasers, fluorescent sensors and organic light-emitting devices.17–19
N–NH– group.3 The hydrazone can be considered for molecular switching due to its inclusion with am imine group that can suffer stimuli responsive cis/trans isomerisation and/or ability to lose proton (Brønsted acid). Hydrazones are usually prepared by three main synthetic pathways: (i) coupling between aryl diazonium salts and β-keto esters or acids active methylene-containing materials, which is also known as the Japp–Klingemann reaction, (ii) condensation of hydrazines and ketones or aldehydes, and (iii) coupling between aryl halides and non-substituted hydrazones.3,4,13–16 On the other hand, DCDHF (2-(dicyanomethylene)-2,5-dihydro-4,5,5-trimethylfuran-3-carbonitrile) chromophores with push-π-pull molecular structure have attracted much attention because of their inherent non-linear optical properties, which are highly sensitive to changes in the environment involving solvent polarity or pH of media, due to their intrinsic character. They have been intensively developed as photo- and electroluminescent materials in many fields including electro-optics, photorefractives, single molecule fluorophores, dye lasers, fluorescent sensors and organic light-emitting devices.17–19
The field of colorimetric spectral signalling involving pH triggered molecular switching has received significant recent attention due to its key role in chemosensing, particularly for environmental monitoring alerts.13,14 A range of analytical techniques has been used for the recognition of ammonia and amines including titrimetric methods, electrochemistry, and UV-Vis absorption spectrometry.20–23 However, these approaches are often not applicable for gas phase recognition, are slow or irreversible, and/or necessitate high operating temperatures. Optical sensing techniques involving absorbing and fluorescent probes have also been used for ammonia and amines. Optical sensors typically present a low cost approach that can be simply formulated from a diverse array miniaturized structures with high sensitivity. Furthermore, colorimetric detection of amines usually employs sensing chromophores comprised of metal-based porphyrins, solvatochromic chromophores, and pH acid/base probes.24–29 Recognition of amines in the solution phase has also been considered broadly. However, sensing of amines in the gas phase has been reported less often as a result of the limitations of sensory materials that permit gas phase recognition with high sensitivity, selectivity and reversibility. Therefore, it is of high significance to design solid state sensors with strong optical recognition, sensitivity and reversibility to gaseous amines.30–32 When deprotonated, a hydrazone moiety can act as a bridge between donor and acceptor fragments in a donor–acceptor system or act itself as a donor fragment when in conjugation with strong electron withdrawing group.33 The p-nitrophenylhydrazone has an acidic N–H group, able to produce hydrazone anion with an enhanced electron donating ability.13,14 Conjugation of this nitrogen hydrazone anion with a strong acceptor group should lead to potentially attractive stimuli-responsive materials.13,14
Herein, we report on synthesis, characterization, and photophysical properties of a novel family of hydrazone chromophores containing DCDHF moiety as a strong acceptor. The polarizability of the system can be improved by replacing the simple phenylhydrazone moiety by a stronger electron withdrawing group such as 4-nitrophenylhydrazone. The spectroscopic properties are explored by electronic absorption and emission spectra and mechanisms of thermochromism and solvatochromism are proposed based on the structural differences. The potential of dye sensors to recognize changes in pH by change of absorption/emission properties is also discussed. Nanostructures with different morphologies were fabricated via a re-precipitation technique from DCDHF-hydrazone (DCDHF-H) chromophores with different substituents. Due to the higher ratio of surface-to-volume, the fabricated miniaturized solid-state films exhibit higher sensitivity upon exposure to a variety of volatile amines. The characteristics and analytical performance of these sensors including reversibility, detection limits, and amine selectivity are explained. Literature techniques were applied to the fabrication of the drop-cast sensor films prepared from 2, 3, and 6.34 In addition, the responsive behaviour of the chromophores 2, 3, and 6 to some selected amines in homogenous solutions are investigated.
Experimental
Materials and methods
Melting points were obtained by differential scanning calorimetry (TA instruments 2920). IR spectra were recorded with a Bruker Vectra-33 IR-spectrometer with ATR Probe. Mass spectra were obtained on a Shimadzu GCMS-QP 1000 EX mass spectrometer at 70 eV. Elemental analyses (C, H, N) were performed on Perkin-Elmer 2400 analyzer (Perkin-Elmer, Norwalk, CT, USA) at the Microanalytical Centre, Cairo University. UV-visible absorption spectra were measured on a HP-8453 (HEWLETT PACKARD) spectrophotometer. Fluorescence emission spectra and quantum yields (Φ) were measured on a VARIAN CARY ECLIPSE spectrophotofluorimeter. NMR spectra were recorded using a BRUKER AVANCE 400 spectrometer at 400 MHz; chemical shifts are given in ppm relative to internal standard TMS at 295 K. The pH measurements were recorded with a BECKMAN COULTER digital pHI340 pH meter, with a combined glass-calomel electrode. Scanning electron microscopy was obtained on Hitachi S-2600N SEM (operating at 20 kV). The SEM samples were prepared by drop-casting the suspension of the obtained nanoparticles, nanorods or nanofibers in hexane on clean glass substrates followed by drying in air. The dried samples were then annealed overnight in an oven at 45 °C, followed by coating with gold. Transmission electron microscopy was performed on JEM-1200EX at an accelerating voltage of 80 kV. All solvents employed were analytically pure and were used without any further purification. All other analytical materials and reagents were purchased from commercial sources and used without further purification. Tetrabutylammonium hydroxide was prepared from tetrabutylammonium iodide according to a literature method.35
pH control for sensors testing
Trifluoroacetic acid was added to solutions of chromophores 2, 3 and 6 to reduce the pH value. The hydrazone anions were produced in situ by addition of a base to solutions of their conjugate acids (DCDHF-H). For example, addition of 1 M methanolic tetrabutylammonium hydroxide to solutions of chromophores 2, 3 and 6 in acetone or dimethyl sulfoxide produced the corresponding hydrazone anions.
Procedure for concentration-dependent 1H NMR measurements
The concentration-dependent 1H NMR was carried out by gradually increasing the concentration of the DMSO-d6 solution of DCDHF-H 2. The initial concentration of 2 is 0.67 mM, the concentration was then adjusted by direct addition of correct quantity of 2 into the DMSO-d6 solution to reach 6.85 mM and (c) 11.38 mM.
Fabrication of nanostructure based film sensors
The nanostructure films based on 2, 3 and 6 were fabricated using a re-precipitation technique.34 Typically, 0.2 mL of concentrated acetone solution of the chromophore (1.0 × 10−3 mol L−1) was injected rapidly into 5.0 mL hexane with vigorous stirring followed by aging for two hours at room temperature. The resulting suspension of nanoparticles, nanofibers and nanorods formed from 2, 3 and 6 respectively were then drop-cast onto glass slides by pipetting and air dried at room temperature.
Sensor testing
A liquid amine (4 mL) was placed in a 10 mL glass vial. After generating amine vapor by heating the vial at the boiling point of respective amine, a glass slide with the nanostructure based film was placed near the top of the vial producing an instant color change. The reversibility of the sensing effect was monitored by the UV-Vis absorption spectra at 517 nm and 592 nm for the chromophore 6 in dimethyl sulfoxide solution (conc. 2.2 × 10−5 mol L−1) at ambient temperature. The absorption spectra were switched back and forth at 517 nm and 592 nm by adding an aqueous solution of ammonia (conc. 2.6 × 10−3 mol L−1) and recording the absorption spectra at 592 nm. This was followed by raising the temperature of the cuvette inside the spectrophotofluorimeter from room temperature to 80 °C and maintaining it for a fixed time (5.0 minutes) to allow the ammonia to completely evaporate. The UV-Vis absorption spectra were recorded again after cooling to room temperature. The same amount of ammonia aqueous solution was added again to monitor the occurrence of the sensing.
Thermal properties
The thermal properties of the chromophores 1–6 were gauged by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (see ESI†). Chromophores 2, 3 and 6, containing nitro electron withdrawing groups on the hydrazone fragment, are all thermally stable up to 226 and 229 °C respectively, as shown by their decomposition temperature (35–45% weight loss temperature) under nitrogen atmosphere in the TGA measurements. On the other hand, chromophores 1, 4 and 5, with electron donating groups on the hydrazone fragment (including aniline based hydrazone; R = H), display a little lower thermal stability up to 222, 208, and 216 °C respectively (8–9% weight loss temperature). DSC experiments were performed by scanning at a rate of 5 °C min−1 under nitrogen atmosphere in the temperature range from 25 to 300 °C. Chromophores 2, 3 and 6 decomposed at 226, 236 and 229 °C respectively, while chromophores 1, 4 and 5 showed slightly lower decomposition temperatures of 217, 215, and 216 °C respectively.
General X-ray crystal structure information
Single crystals of compound 4 suitable for X-ray diffraction analysis were obtained by crystallization (with slow solvent evaporation for 24 hours at room temperature) from a mixed solvent (ethanol/toluene, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Crystal data were collected by mounting a crystal onto a thin glass fibre from a pool of Fluorolube™ and immediately placing it under a liquid N2 cooled stream, on a Bruker AXS diffractometer upgraded with an APEX II CCD detector. The radiation used is graphite monochromatized Mo Kα radiation (λ = 0.7107 Å). The lattice parameters are optimized from a least-squares calculation on carefully centered reflections. Lattice determination, data collection, structure refinement, scaling, and data reduction were carried out using APEX2 Version 2014.11 software package. The data were corrected for absorption using the SCALE program within the APEX2 software package. The structure was solved using direct methods. This procedure yielded a number of the C and N atoms. Subsequent Fourier synthesis yielded the remaining atom positions. The hydrogen atoms are fixed in positions of ideal geometry (riding model) and refined within the XSHELL software package. These idealized hydrogen atoms had their isotropic temperature factors fixed at 1.2 or 1.5 times the equivalent isotropic U of the C atoms to which they were bonded. A few hydrogen atoms could not be adequately predicted via the riding model within the XSHELL software. These hydrogen atoms were located via difference-Fourier mapping and subsequently refined. The final refinement of each compound included anisotropic thermal parameters on all non-hydrogen atoms. Crystallographic data (excluding structure factors) for the structure in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1487705.
:1). Crystal data were collected by mounting a crystal onto a thin glass fibre from a pool of Fluorolube™ and immediately placing it under a liquid N2 cooled stream, on a Bruker AXS diffractometer upgraded with an APEX II CCD detector. The radiation used is graphite monochromatized Mo Kα radiation (λ = 0.7107 Å). The lattice parameters are optimized from a least-squares calculation on carefully centered reflections. Lattice determination, data collection, structure refinement, scaling, and data reduction were carried out using APEX2 Version 2014.11 software package. The data were corrected for absorption using the SCALE program within the APEX2 software package. The structure was solved using direct methods. This procedure yielded a number of the C and N atoms. Subsequent Fourier synthesis yielded the remaining atom positions. The hydrogen atoms are fixed in positions of ideal geometry (riding model) and refined within the XSHELL software package. These idealized hydrogen atoms had their isotropic temperature factors fixed at 1.2 or 1.5 times the equivalent isotropic U of the C atoms to which they were bonded. A few hydrogen atoms could not be adequately predicted via the riding model within the XSHELL software. These hydrogen atoms were located via difference-Fourier mapping and subsequently refined. The final refinement of each compound included anisotropic thermal parameters on all non-hydrogen atoms. Crystallographic data (excluding structure factors) for the structure in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1487705.
Results and discussion
Synthesis and characterization
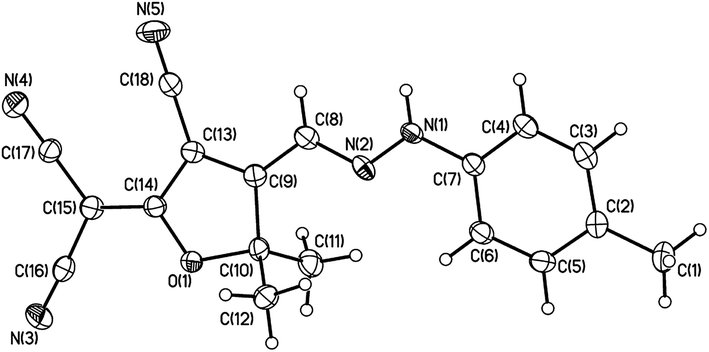
The simple synthetic approach for the DCDHF-H chromophores 1–6 is shown in Scheme 1. The intermediate 2-(dicyanomethylene)-2,5-dihydro-4,5,5-trimethylfuran-3-carbonitrile 7 was prepared according to the literature method.13,14 It will be helpful to describe some of the related pieces of the proposed chemistry and clarify their relation with the existing goal of our novel generation of DCDHF-H chromophores. Knoevenagel condensation reaction has been important in the production of electron deficient head materials such as the high electron withdrawing DCDHF heterocycle head that are well-known in push–pull systems (ESI†).14 In a broad variety of reactions, the active methylene materials such as malononitrile or α-nitrile bearing carbonyls can react with bases such as piperidine, sodium ethoxide, and pyridine in order to couple with diazonium salts or condense with ketones or aldehydes. Subsequent reaction with an extra equivalent of active methylene compound followed by intramolecular ring closure leads to highly functionalized electron deficient heterocycles in one pot. The hydrogen atoms on a carbon adjacent to highly electron withdrawing groups in a molecule are considerably more acidic than hydrogen atoms on a carbon adjacent to alkyl units.14 Here, the electron withdrawing groups helps stabilize the carbanion produced from the removal of a hydrogen proton from the activated methyl or methylene group using sodium acetate. The degree of resonance stabilization of this DCDHF's conjugate anion is depicted by the proposed six resonance structures (ESI†). The preparation was simply performed via azo-coupling starting from 2-(dicyanomethylene)-2,5-dihydro-4,5,5-trimethylfuran-3-carbonitrile and the respective aryl diazonium chloride derivatives. The azo coupling process is base-mediated reaction between the diazonium salt and DCDHF head bearing active methyl group to afford the corresponding unstable azo intermediate that transform directly to the more stable hydrazone form. The hydrazone NH functional group was confirmed by 1H NMR in the range 12.11–12.80 ppm; and by FT-IR represented through a peak between 3241 and 3391 cm−1. The spatial structure of compound 4 in the hydrazone form was proved by single crystal X-ray crystallography as shown in Fig. 1. The molecule displays an E conformation with respect to the C(8)N(2) double bond, with a C(9)–C(8)–N(2)–N(1) torsion angle of −179.29°. Bond lengths and angles are unexceptional and similar to those found in related structures.36 The N(1)–N(2) bond length is 1.311 Å shorter than the usual hydrazine N–N bond length (1.38 Å), but is closer to other similar molecules containing the N–N moiety, such as dihydrazones.37 The C(8)N(2) double bond length is 1.313 Å shorter than the N(1)–C(7) single bond length is 1.411.
 |
| | Scheme 1 Synthesis of DCDHF-hydrazone chromophores 1–6. | |
 |
| | Fig. 1 Crystal structure (ORTEP view) of compound 4 (R = 4-CH3) as derives from the X-ray analysis (thermal ellipsoid plots are drawn at the 30% probability level). | |
Thermal properties
The thermal properties of the chromophores 1–6 were gauged by thermogravimetric analysis and differential scanning calorimetry (ESI†). Chromophores 2, 3 and 6, containing nitro electron withdrawing groups on the hydrazone fragment, are all thermally stable up to 226 and 229 °C respectively, as shown by their decomposition temperature (35–45% weight loss temperature) under nitrogen atmosphere in the TGA measurements. On the other hand, chromophores 1, 4 and 5, with electron donating groups on the hydrazone fragment (including aniline based hydrazone; R = H), display a little lower thermal stability up to 222, 208, and 216 °C respectively (8–9% weight loss temperature). DSC experiments were performed by scanning at a rate of 5 °C min−1 under nitrogen atmosphere in the temperature range from 25 to 300 °C. Chromophores 2, 3 and 6 decomposed at 226, 236 and 229 °C respectively, while chromophores 1, 4 and 5 showed slightly lower decomposition temperatures of 217, 215, and 216 °C respectively.
Photophysical properties
Solvatochromism and solvatofluorochromism. The UV-Vis absorption and fluorescence spectra of the DCDHF-based aryl-hydrazone chromophores 1–6 in a variety of solvents with different polarities (at fixed neutral pH value) are shown in Table 1S.† The colours of these DCDHF-H chromophores in various pure solvents range from yellow to purple. Inspection of Table 1S† indicates that the longest wavelength λmax value of chromophores 1–6 in the same solvent increases in the order 3 < 2 < 6 < 1 < 4 < 5. Replacement of the parent phenyl-hydrazone by the more polarizable 4-nitrophenyl-hydrazone moiety caused a larger bathochromic shift of this band. In general, the maximum absorption peak undergoes a bathochromic shift (positive solvatochromism) with increasing solvent polarity (ca. 155 nm as the maximum shift value for R = 2,4-di-NO2 and ca. 24 nm as the minimum shift value for R = H; on going from chloroform to benzonitrile). These features indicate a strongly allowed π–π* transition with charge transfer character. For chromophores 2, 3 and 6, distinct solvatochromic activity was observed in polar protic and aprotic solvents. In protic solvents, a discernible blue shift resulted from increasing the acidity of the alcoholic solvent. In aprotic solvents, the λmax values for a given chromophore were generally larger than in alcoholic solvents. A larger range in the maximal λmax values was observed for the nitro-derivatives 2, 3 and 6, with little consistent variation of the transition energies with the medium polarity. The nitro-substituted DCDHF-H chromophores 2, 3 and 6, reveal a higher acidity of the hydrazone NH than chromophores 1, 4 and 5 and the dinitro-substituted chromophore 3 indicates a still stronger effect than the mono-substituted 2 and 6. These strong electron withdrawing groups enhance the stability of the hydrazone anion. The solvatochromic behaviour can be explained by two extreme resonance structural contributions. One form is quinoidal, nonpolarized, and not fully aromatic; while the other form is zwitterionic, polarized, and fully aromatic. The variation in the absorption peak with changing solvent polarity arises from variation in the contribution extent of these canonical forms to the overall resonance hybrid. Hence, the nitro-groups certainly play a significant role in promoting solvatochromism. An intramolecular charge transfer from the hydrazone donor moiety to the DCDHF acceptor fragment is induced upon excitation as indicated by the bathochromic shift of the fluorescence maximum in polar solvents. The solvent dependence of the fluorescence spectra is termed solvatofluorochromism. The emission maximum wavelength of the DCDHF-H chromophore (3; R = 2,4-diNO2) is bathochromically shifted ca. 152 nm representing the maximum shift value, while the DCDHF-H chromophore (1; R = H) represents the minimum shift value ca. 27 nm; with changing the solvent from 1,4-dioxane to benzonitrile. Such a behaviour indicates stabilization of the highly dipolar excited state in polar solvents. DCDHF-H molecules 1–6 exhibit relatively high molar absorptivity coefficient (ε) and low fluorescence quantum yields (Φ) in all solvents. Due to the low quantum yield and very low solubility in n-propanol compared to ethanol and methanol; no fluorescence is detected for compounds 1, 4, 5 and 6. On the other hand, the highly polar DMF solvent has a little alkaline character and ability to deprotonate the hydrazone form into the hydrazone-anion form which causes a significant increase in the charge density on the hydrazone nitrogen with an associated increase of the intramolecular charge transfer and consequently no fluorescence is detected for the compounds 1–6.It was monitored that compound 3 (R = 2,4-dinitro) exhibits dual absorption/dual emission in DMSO at neutral pH value (Table 3S†). At room temperature, the excitation spectrum of chromophore 3 is characterized by two main absorption bands in the visible range at 467 nm for the hydrazone form (S-2-H) and 594 nm for the hydrazone anion form (S-2-HA) which is attributed to an internal ground-state equilibrium between the neutral (S-2-H) and anionic (S-2-HA) hydrazone forms, respectively. The excited state of the neutral chromophore fluoresces at 551 nm and the anionic chromophore at 668 nm. These two absorption bands is a result of different protonation states, S-2-H and S-2-HA, with the high-energy band corresponding to the neutral chromophore and the low energy band to the anionic chromophore. Furthermore, a Twisted Internal Charge Transfer (TICT) causes the highly electron withdrawing effect of two nitro substituents, on the aromatic ring of the hydrazone fragment, in the most polar excited state of S-2-HA to be markedly twisted in the highly polar DMSO solvent out of the molecular plane. Therefore, the anionic form of S-2-HA containing highly electron withdrawing effect of two nitro groups is fluorescence active in the highly polar DMSO solvent.38,39
pH-induced molecular switching. The sensing potential of the nitro-substituted DCDHF-H chromophores 2, 3 and 6, to undergo molecular switching imparted by pH stimulus was established by the addition of base and acid to a 2–3 × 10−5 M solution of DCDHF-H (2, 3 and 6) in acetone or dimethylsulfoxide. Fig. 2 shows the reversible UV-Vis electronic absorption spectra and colour switching changes of DCDHF-H chromophore 2 in acetone, with protonation and deprotonation. Upon the addition of tetrabutylammonium hydroxide base to the solution of chromophore 2 in acetone, the peak of maximal wavelength λmax at 462 nm decreased and a new peak at 558 nm appeared; the addition of trifluoroacetic acid then resulted in the absorption peak at 462 nm to reappear (Fig. 2). Thus, fluorescence emission intensity was varied, reversibly, at 557 nm (excitation at 488 nm in dimethylsulfoxide solution) by the alternate addition of tetrabutylammonium hydroxide and trifluoroacetic acid (Fig. 3). Upon the addition of tetrabutylammonium hydroxide, the fluorescence emission peak at 557 nm progressively decreases, while the same peak intensity increases with the addition of trifluoroacetic acid. The property of pH-sensitivity is accomplished by incorporating a nitro-based aryl-hydrazone as a donor fragment. Upon protonation of the hydrazone nitrogen, fluorescence emission intensity increases significantly. The presence of a distinct isosbestic point at 501 nm indicates that only two species coexist in equilibrium and these spectral variations are due to the high acidity of the nitro-substituted chromophores. This suggests that a negatively charged hydrazone anion was generated in basic medium, which causes a significant increase in the charge density on the hydrazone nitrogen with an associated increase of the intramolecular charge transfer (Fig. 4). The net result is that the electronic transition energy is lower for the hydrazone anion form.
 |
| | Fig. 2 (a) The pH dependent absorption spectra; (b) the colour changes (yellow, orange, orange-red, wine-red, violet, and purple at pH 5.14, 5.30, 5.39, 5.84, 6.07, and 6.19 respectively); of DCDHF-H (2; R = 4-NO2) in acetone solution (conc. 2.2 × 10−5 mol L−1) at different pH values at ambient temperature. | |
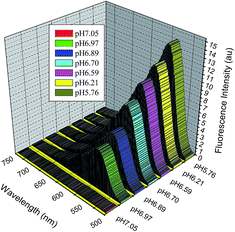 |
| | Fig. 3 Changes of the fluorescence emission spectra of 2 (excitation wavelength at 488 nm) in dimethylsulfoxide solution (conc. 2.5 × 10−5 mol L−1) at different pH values at ambient temperature. | |
 |
| | Fig. 4 The pH of the environment has a dramatic influence on the electronic properties of the chromophores and their respective absorption wavelengths. | |
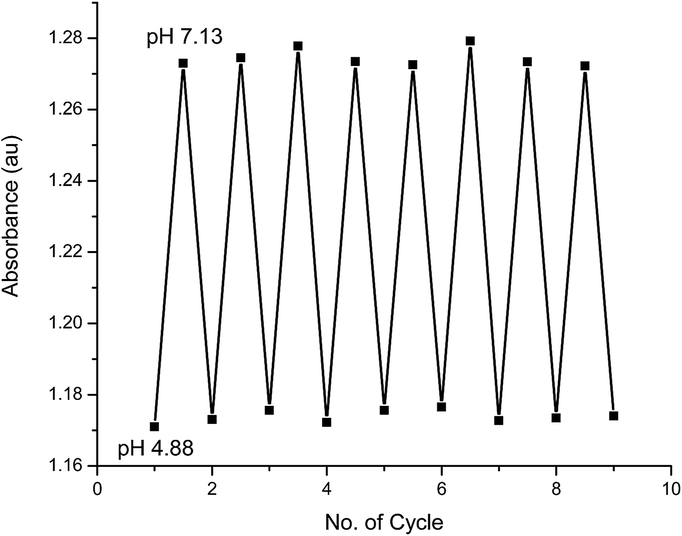
To examine the reversibility of these pH-dependent variations, 1 M methanolic solutions of tetrabutylammonium hydroxide and trifluoroacetic acid were used to change the pH back and forth between 4.88 and 7.13; which was monitored by pH meter. The ratios of the absorbance at 462 and 558 nm of chromophore 2 were measured and the results are shown in Fig. 5. It is clear that this process has a high reversibility showing that compound 2 has a high stability under these conditions.
 |
| | Fig. 5 Changes in the ratio of the absorbance at 462 and 558 nm of 2 in acetone solution (conc. 2.2 × 10−5 mol L−1) at different pH values at ambient temperature. The pH value was switched back and forth between 4.88 and 7.13 using tetrabutylammonium hydroxide and trifluoroacetic acid. | |
Thermochromic behaviour. The yellow colour of the highly sensitive dinitro-substituted DCDHF-H chromophore 3 changed to purple upon heating in dimethylsulfoxide solution. The reversible colour changes are easily observed with chromophores 2, 3 and 6 upon heating (Fig. 6). However, no similar colour change was observed with chromophores 1, 4 and 5. Obviously, the nitro group is necessary to thermochromism of DCDHF-H. The induced planarity of the nitro-substituted DCDHF-H chromophores are better than those with electron donating groups, on the other hand, the two proposed isomeric molecular species of nitro-substituted DCDHF-H chromophores exhibit a molecular switching behaviour upon heating. The thermochromism of organic compounds takes place via an equilibrium progression between two isomeric molecular canonical species. The extended conjugation of the hydrazone anion structure is apparently larger than that of the neutral chromophore, which is favourable to the electron transfer, so the colour changes from yellow to purple. As a matter of fact, the tautomerism of the dinitro-substituted DCDHF-H chromophore 3 can be proved by the fluorescence emission spectra in dimethylsulfoxide solution (conc. 2.9 × 10−5 mol L−1) at various temperatures (20–95 °C). The emission intensities at both 551 nm and 667 nm were gradually reduced and had an 8% and 23% value (at 95 °C) of their primary values at 20 °C.
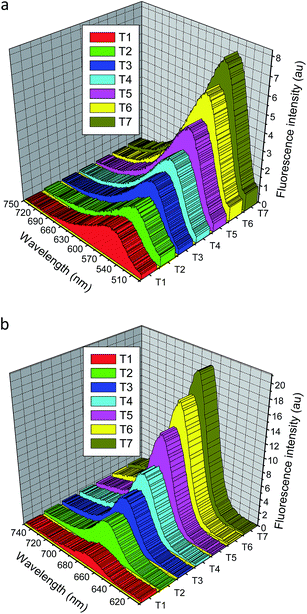 |
| | Fig. 6 Fluorescence emission intensity of the dinitro-substituted DCDHF-H chromophore 3; (a) at excitation wavelength 467 nm, and (b) at excitation wavelength 594 nm; in dimethylsulfoxide solution (conc. 2.9 × 10−5 mol L−1) at various temperatures (T1 = 95 °C, T2 = 85 °C, T3 = 75 °C, T4 = 65 °C, T5 = 50 °C, T6 = 35 °C, and T7 = 20 °C). | |
The results suggest that a strong quinoid-type interaction exists between the 4-nitrophenyl group and the lone-pair electrons on the amino nitrogen atom in the hydrazone fragment, while such an extra resonance effect is depressed in the hydrazone anion presumably because the negative charge interacts instead more strongly to the strong DCDHF acceptor fragment. The 4-nitro-substituted DCDHF-H chromophore 2 can exist in two distinct resonance forms: a neutral molecule and a zwitterion. The zwitterionic form dominates in polar solvents, and the neutral quinoid form dominates in nonpolar solvents. The 4-nitro-substituted DCDHF-H chromophore and the equivalent resonance structures of its hydrazone anion are shown in Fig. 7S.† The deprotonated HA form was also confirmed by both of IR and 1H NMR spectra for DCDHF-H 2. The characteristic secondary hydrazone NH band (at 3274 cm−1) was disappeared in the IR spectra of 2 extracted from a mixture of acetonitrile/triethylamine. The 1H NMR spectra was reported at both room temperature and 90 °C in DMSO-d6 solution of 2. The NH peak (at 12.80 ppm) was disappeared in the 1H NMR spectra of 2 upon increasing temperature (ESI†).
Detection of ammonia and amines in aqueous solution. There is an increasing demand for the sensing of amines in both gaseous and solution phases. The presence of amines in solution affects the pH of the medium. The interaction of the DCDHF-H sensory chromophores with aliphatic and aromatic amines in organic solutions is investigated. The UV-Vis absorption spectra of the employed sensors 2, 3 and 6 were monitored upon addition of increasing concentrations of aqueous ammonia (Fig. 7). When reacting with amines, the hydrazone N–H group is reversibly converted by deprotonation into a hydrazone anion (HA).14 This acid–base reaction is accompanied by an increase in the electron donating effect of the hydrazone anion (HA) group, the electron delocalization within the chromophore molecule is modulated and, therefore, the absorption maxima resulting from the deprotonated molecule is shifted to longer wavelength. To study the hydrazone anion formation, a wide variety of amines with different pKa values (amines 1–20; Fig. 8S†) including aromatic and aliphatic (primary, secondary, tertiary, cyclic/acyclic and mono/diamine) amines. The type of amine used did not significantly influence the essential optical properties of the hydrazone anion formed. The UV-Vis absorption and fluorescence spectra of each sensor and its corresponding HA form in some selected solvents are shown in Table 3S.† The stepwise addition of amines to a variety of organic solutions of 2, 3 and 6 and subsequent recording of the corresponding absorption maxima λmax in acetone led to a series of spectra with one isosbestic point for the sensors 2, 3 and 6 at 502 nm, 498 nm, and 519 nm respectively (Fig. 7a and b).
 |
| | Fig. 7 The UV-Vis absorption spectra of sensors 2 (S-1-H) in acetone (a), 3 (S-2-H) in acetone (b), and 6 (S-3-H) in ethanol (c); upon addition of increasing concentrations of ammonium hydroxide aqueous solution. | |
As a result of hydrazone anion formation, the intensity of the absorption at shorter wavelength decreased while a simultaneous longer wavelength absorption appeared and grew in intensity. The resulting absorption spectra indicated a significant bathochromic shift between the neutral DCDHF-H and the hydrazone anion derivative with increasing number of the strong nitro electron withdrawing groups in the sensor dye. As a result of the electron donating OH group at the ortho position; the shorter wavelength maxima at 461 nm of 6 upon addition of amines and the absorption band of the corresponding hydrazone anion form have a smaller wavelength shift difference to longer wavelength than 2 and 3. Interestingly, each of the three sensors in protic solvents (such as ethanol) displays a series of spectra with two isosbestic points for each chromophore (at 382/504 nm, 377/499 nm, and 397/524 nm respectively (Fig. 7c). One explanation for this additional isosbestic point appearing in protic solvents involves hydrogen bonding between the hydrazone anion and ethanol as the bands at 382 nm, 377 nm, and 397 nm respectively; increases with increasing the corresponding hydrazone anion bands at 504 nm, 499 nm, and 524 nm respectively. This suggests that the hydrogen bonding between the hydrazone anion and ethanol leads to a new species in equilibrium with other two species; the hydrazone dye and its corresponding hydrazone anion (Fig. 9S†).14 The fluorescence wavelength λmax maxima of the sensory dyes were recorded with the stepwise addition of amines in a variety of solvents. In contrast to the unreacted (protonated) DCDHF-H derivatives, the corresponding hydrazone anions did not show any fluorescence in the selected solvents. While the amount of amine added to the dye solution is increased, the fluorescence intensity decreases in proportion, which indicates the formation of the corresponding hydrazone anion.
All amines react instantly to form the hydrazone anion (HA) that leads to the quinoidal form. The changes in absorption spectra (around 580 nm) due to addition of a variety of amines to acetone solutions containing the molecular probe 6 are depicted in Fig. 8 and Table 4S.† The response and sensitivity of 3 towards amines is higher than that found for 2 and 6. The reason for the difference in sensitivity is based on the difference in their chemical structures as a result of the two strong nitro electron withdrawing groups. Hence, in the case of the reaction of 2 or 6 with amines leads to low acidity of the hydrazone NH group and a lower possibility to form the hydrazone anion than in the case of 3, which exhibits higher acidity of the hydrazone NH group. It is found that chromophores exhibit selective sensitivity for different organic amines as the amines basicity is the key controller of sensing effect. Generally, the UV-Vis absorption spectra are almost compatible with the amines pKb values. The sensor dyes show comparable sensitivity for n-ethylamine, diethylamine and triethylamine, with highest sensitivity for the secondary amines and less sensitivity for the primary and then tertiary amines. Less basic aromatic and heterocyclic amines such as aniline and pyridine proved more difficult to detect. Distinct selectivity is also observed along each series of the employed amines, parallel to the basicity of each amine.
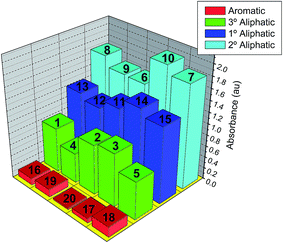 |
| | Fig. 8 Changes in absorption maxima of the 6 hydrazone-anion form (at 580 nm) in acetone solution containing the molecular probe 6 hydrazone form after addition of different types of amines (4.0 μM). | |
The reversibility of the sensing effect towards amines was monitored by warming the chromophore solution in the cuvette in situ from room temperature to 80 °C and maintaining it for a fixed time (5.0 minutes) to allow the amine to evaporate. The UV-Vis electronic absorption and fluorescence emission spectra were recorded again after cooling to room temperature. The same amount of ammonium hydroxide aqueous solution was added again to monitor the reoccurrence of the sensing. The changes in the ratio of the absorbance at 517 nm and 592 nm of 6 in dimethyl sulfoxide solution (conc. 2.2 × 10−5 mol L−1) were recorded at ambient temperature. The absorbance values were switched back and forth between 2.19 and 2.31 respectively adding ammonium hydroxide aqueous solution (conc. 2.6 × 10−3 mol L−1) followed by heating. As shown in Fig. 10S†, at least through this many cycles it is clear that this process exhibits high reversibility without fatigue towards amine sensing.
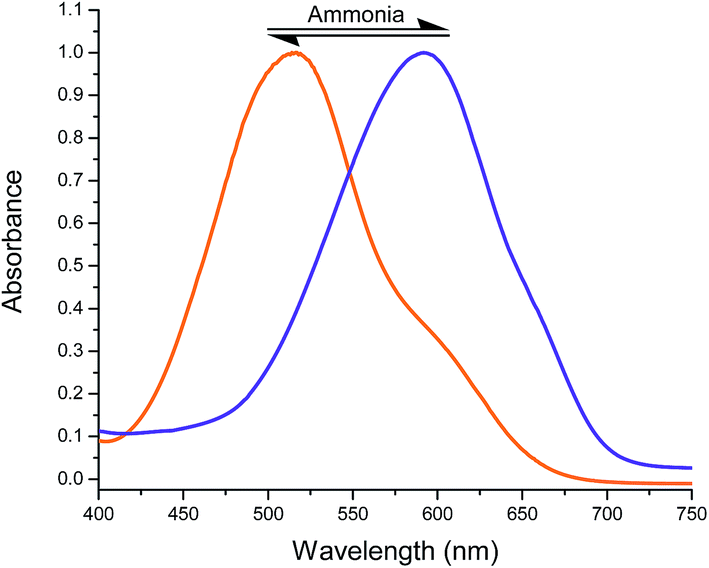
Colorimetric sensing of ammonia and amines vapours. Nanostructures assembled from DCDHF-based chromophore with a hydrazone moiety in the backbone function as vapochromic colorimetric sensor toward volatile alkaline molecules. These nanostructures were fabricated using a re-precipitation technique followed by drop-casting the dye solution in hexane onto a glass substrate. The solid state film samples produced after evaporation of the hexane were exposed to amine vapours displaying an instant colour change from orange to purple, which was monitored by naked eye (Fig. 9). Upon removal of the film sensor away from the amine vapour, the sample quickly regains its previous orange colour as the amine vapour concentration diminishes. The sensor could be used repeatedly with a variety of amines without any apparent degradation. The morphology of the nanostructures fabricated from sensors 2, 3 and 6 through a rapid solution dispersion process were investigated using both of scanning electron microscopy and transmission electron microscopy. From Fig. 10, we found three dimensional networks consisting of nanoparticles (fabricated from 2) with width of ca. 100–250 nm, nanofibers (fabricated from 3) with width of 200–350 nm and few micrometers in length or nanorods (fabricated from 6) with width of 250–450 nm and few micrometers in length.
 |
| | Fig. 9 Normalized UV/Vis absorption spectra of nanofibers from S-2-H (R = 2,4-diNO2); before (orange; at 507 nm) and after (blue; at 584 nm) exposure to ammonia vapours. | |
 |
| | Fig. 10 Scanning electron microscope and transmission electron microscope images (a and d respectively) displays nanoparticles fabricated from 2, (b and e respectively) displays nanorods fabricated from 6, and (c and f respectively) displays nanofibers fabricated from 3; indicating different morphologies were fabricated from the DCDHF-based molecular switches with different substituents; for solid-state reversible optical sensing of amines in gaseous phase. | |
It is anticipated that these thinner solid state nanostructure based sensors may exhibit higher sensitivity in the detection of amines due to higher ratio of surface-to-volume, and larger interspaced networks enabling the diffusion of guest amines through the matrix. The nanostructure based films derived from 2, 3 and 6 were fabricated using a re-precipitation technique. Typically, 0.2 mL of concentrated acetone solution of the dye (1 × 10−3 mol L−1) was injected rapidly into 5.0 mL of vigorously stirred hexane followed by aging for two hours at room temperature to give nanoparticles, nanofibers and nanorods formed from 2, 3 and 6, respectively. The suspension of nanoparticles, nanorods and nanofibers thus prepared in hexane can be drop-cast onto glass slides by pipetting then air dried at room temperature. The diameter of the produced nanostructures (nanoparticles, nanofibers and nanorods) based on 2, 3 and 6 was ca. 180 nm, ca. 250 nm and ca. 350 nm on average, respectively. To test the film sensor a liquid amine (4.0 mL) was placed in a 10.0 mL glass vial. After generating amine vapor by heating the vial at the boiling point of respective amine, the nanostructure based film was placed near the top of the vial demonstrating instant color change.
Morphology and structure of assemblies. It is of interest that the substituents on the hydrazone moiety play an important role in the self-assembly process and lead to nanostructures with different morphologies depending on the substituents on the phenyl hydrazone moiety. In the concentration-dependent 1H NMR as shown in Fig. 11, the N–H resonance signals showed apparent downfield shifts as the concentration of 2 increased. Similar behaviour was monitored for the aliphatic C–H resonance signals displayed apparent downfield shifts as the concentration of 2 increased. These results revealed that both of N–H and aliphatic C–H groups contributed in the formation of hydrogen bonding in the self-assembly process into the nanostructures. Therefore, it is only the neutral hydrazone form that has any propensity for self-assembly. On the other hand, we were not able to produce self-assembled nanostructures from the hydrazone-anion form due to losing the hydrogen bonding of the N–H group after deprotonation.
 |
| | Fig. 11 Partial 1H NMR spectra of DCDHF-H 2 in DMSO-d6 at different concentrations (a) 0.67 mM; (b) 6.85 mM; and (c) 11.38 mM. | |
Conclusions
We have investigated the photophysical and electrochemical properties of novel hydrazone type DCDHF-based chromophores. These dyes exhibited both positive solvatochromic and solvatofluorochromic properties in solvents with different polarities. The products exhibit reversible colour changes responding to different pH as a result of extensive charge delocalization in the hydrazone-form that leads to a quinoid-type interaction. The induced planarity of DCDHF-H is better in systems possessing strong electron withdrawing groups leading to larger conjugation degree of the formed hydrazone anion than that of the neutral chromophore, which is favourable to the intramolecular charge transfer. An examination of the UV-Vis spectra investigation indicates that DCDHF-H is solvatochromic. The colour of DCDHF-H changes from yellow to red and blue, furthermore, the colors of DCDHF-NH are diverse in different pure solvents at the same conditions. A pH dependent molecular switching was demonstrated by modulation of intramolecular charge transfer with protonation and deprotonation. The hydrazone recognition moieties are employed for the detection of amines in the solution phase at nanomolar concentration levels using UV-Vis electronic absorption and fluorescence emission spectroscopy. The nanostructures fabricated from DCDHF-H dyes were shown to act as a reversible colorimetric sensor for amine vapours. The nanostructure aggregates of DCDHF-hydrazones exhibit solid-state optical sensory for vapour sensing, together with the specific active hydrazone anion site offered by the NH functional group acidity. Exposure to common amines vapours led to a change of the observed colour towards purple. The amine basicity is the key controller of detection. It is found that these chromophores exhibit selective sensitivity for different organic amines. Less basic aromatic and conjugated amines such as pyridine, and aniline are less readily detected while secondary aliphatic amines are the most readily detected. The current approach introduces a simple and appropriate technique to rank the relative Lewis basicity of different organic amines.
Acknowledgements
This research was supported by the National Science Foundation through the Center for Layered Polymer Systems (CLiPS) under Grant No. 0423914. This material was also supported by the USDA grant # 58-3148-2-113.
References
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- Y.-S. Mi, D.-M. Liang, Y.-T. Chen, X.-B. Luo and J.-N. Xiang, RSC Adv., 2014, 4, 42337–42345 RSC.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- I. Aprahamian, Nat. Chem., 2016, 8, 97–99 CAS.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed.
- F.-F. Tian, J.-H. Li, F.-L. Jiang, X.-L. Han, C. Xiang, Y.-S. Ge, L.-L. Li and Y. Liu, RSC Adv., 2012, 2, 501–513 RSC.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- H. Zollinger, Color chemistry: syntheses, properties, and applications of organic dyes and pigments, John Wiley & Sons, 2003 Search PubMed.
- H. Qian and I. Aprahamian, Chem. Commun., 2015, 51, 11158–11161 RSC.
- C. Serbutoviez, C. Bosshard, G. Knoepfle, P. Wyss, P. Pretre, P. Guenter, K. Schenk, E. Solari and G. Chapuis, Chem. Mater., 1995, 7, 1198–1206 CrossRef CAS.
- R. Lygaitis, V. Getautis and J. V. Grazulevicius, Chem. Soc. Rev., 2008, 37, 770–788 RSC.
- T. A. Khattab, S. Abdelmoez and T. M. Klapötke, Chem.–Eur. J., 2016, 22, 4157–4163 CrossRef CAS PubMed.
- T. A. Khattab, B. D. B. Tiu, S. Adas, S. D. Bunge and R. C. Advincula, Dyes Pigm., 2016, 130327–130336 Search PubMed.
- P. Ball and C. H. Nicholls, Dyes Pigm., 1982, 3, 5–26 CrossRef CAS.
- L. Wang, D.-G. Guo, Y.-Y. Wang and C.-Z. Zheng, RSC Adv., 2014, 4, 58895–58901 RSC.
- S. J. Lord, N. R. Conley, H. L. D. Lee, S. Y. Nishimura, A. K. Pomerantz, K. A. Willets, Z. Lu, H. Wang, N. Liu, R. Samuel and R. Weber, ChemPhysChem, 2009, 10, 55–65 CrossRef CAS PubMed.
- Z. Lu, N. Liu, S. J. Lord, S. D. Bunge, W. E. Moerner and R. J. Twieg, Chem. Mater., 2009, 21, 797–810 CrossRef CAS PubMed.
- R. Twieg, H. Wang, Z. Lu, S. Y. Kim, S. Lord, S. Nishimura, P. J. Schuck, K. A. Willets and W. E. Moerne, Nonlinear Opt., Quantum Opt., 2006, 34, 241 Search PubMed.
- N. Chartuprayoon, C. M. Hangarter, Y. Rheem, H. Jung and N. V. Myung, J. Phys. Chem. C, 2010, 114, 11103–11108 CAS.
- Z. M. Siddiqi and D. Pathania, Talanta, 2003, 60, 1197–1203 CrossRef CAS PubMed.
- I. S. Park, E. Heo, Y. S. Nam, C. W. Lee and J. M. Kim, J. Photochem. Photobiol., A, 2012, 238, 1–6 CrossRef CAS.
- Y. Che, X. Yang, Z. Zhang, J. Zuo, J. S. Moore and L. Zang, Chem. Commun., 2010, 46, 4127–4129 RSC.
- Y. Takagai, Y. Nojiri, T. Takase, W. L. Hinze, M. Butsugan and S. Igarashi, Analyst, 2010, 135, 1417–1425 RSC.
- W. Qin, P. Parzuchowski, W. Zhang and M. E. Meyerhoff, Anal. Chem., 2003, 75, 332–340 CrossRef CAS PubMed.
- T. Mizutani, K. Wada and S. Kitagawa, J. Org. Chem., 2000, 65, 6097–6106 CrossRef CAS PubMed.
- Y. Che, X. Yang, S. Loser and L. Zang, Nano Lett., 2008, 8, 2219–2223 CrossRef CAS PubMed.
- J. Kim, S. H. Lim, Y. Yoon, T. D. Thangadurai and S. A. Yoon, Tetrahedron Lett., 2011, 52, 2645–2648 CrossRef CAS.
- X. Zhang, X. Liu, R. Lu, H. Zhang and P. Gong, J. Mater. Chem., 2012, 22, 1167–1172 RSC.
- G. Giancane, V. Borovkov, Y. Inoue and L. Valli, J. Colloid Interface Sci., 2012, 385, 282–284 CrossRef CAS PubMed.
- H. Liu, J. Mack, Q. Guo, H. Lu, N. Kobayashi and Z. A. Shen, Chem. Commun., 2011, 47, 12092–12094 RSC.
- J. J. Peterson, A. R. Davis, M. Werre, E. B. Coughlin and K. R. Carter, ACS Appl. Mater. Interfaces, 2011, 3, 1796–1799 CAS.
- D. Millán, M. Domínguez and M. C. Rezende, Dyes Pigm., 2008, 77, 441–445 CrossRef.
- X. Liu, X. Zhang, R. Lu, P. Xue, D. Xu and H. Zhou, J. Mater. Chem., 2011, 21, 8756–8765 RSC.
- I. Y. Ponedel'kina, E. V. Sal'nikova, E. S. Lukina, T. V. Tyumkina and V. N. Odinokov, Chem. Nat. Compd., 2012, 1–5 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- B. G. Rao and U. C. Singh, J. Am. Chem. Soc., 1991, 113, 4381–4389 CrossRef CAS.
- J. Catalán, Phys. Chem. Chem. Phys., 2013, 15, 8811–8820 RSC.
- G. T. Hanson, T. B. McAnaney, E. S. Park, M. E. P. Rendell, D. K. Yarbrough, S. Chu, L. Xi, S. G. Boxer, M. H. Montrose and S. J. Remington, Biochemistry, 2002, 41, 15477–15488 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental section including description of the synthesis and characterization of the compounds; 1H and 13C NMR, IR, elemental analysis, SEM, TEM, X-ray crystal data and video abstract. The X-ray crystallographic files in CIF format for compound 4. CCDC 1487705. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra24113a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Sonya Adasad,
Scott D. Bungea and
Rigoberto C. Advinculab
b,
Sonya Adasad,
Scott D. Bungea and
Rigoberto C. Advinculab