
A nano-organo catalyzed route towards the efficient synthesis of benzo[b]pyran derivatives under ultrasonic irradiation†
Ridaphun Nongrumab,
Geetmani Singh Nongthombamb,
Mattilang Kharkongorc,
Jims World Star Ranib,
Noimur Rahmanb,
Chingrishon Kathingb,
Bekington Myrbohb and
Rishanlang Nongkhlaw*b
aDepartment of Chemistry, Sankardev College, Bishnupur, Shillong, Meghalaya-793004, India
bCentre for Advanced Studies in Chemistry, North-Eastern Hill University, Shillong, Meghalaya-793022, India. E-mail: rlnongkhlaw@nehu.ac.in
cDepartment of Zoology, Sankardev College, Bishnupur, Shillong, Meghalaya-793004, India
First published on 8th November 2016
Abstract
An efficient, one-pot synthesis of benzo[b]pyrans catalyzed by vitamin B1 functionalized Fe2O3@SiO2 NPs under ultrasonic conditions is reported herein. The nano-organocatalyst was synthesized and characterized by FT-IR, TEM, EDS, powder XRD, etc. Several benzo[b]pyran derivatives were prepared with satisfactory yields and the nanocatalyst was magnetically retrieved and recycled. The salient features of this protocol include convenient work-up procedure, mild reaction conditions, easy catalyst recovery, shorter reaction time and use of green solvents. Ultrasound energy has been meticulously exploited for the synthesis of a nano-organocatalyst as well as for the preparation of several benzo[b]pyran derivatives. The biological activity of the synthesized benzo[b]pyrans were investigated and one of the derivatives was found to demonstrate profound activity in suppressing the side-effects of chlorpyrifos, an insecticide on the morphology of the external gills of the Duttaphrynus melanostictus tadpoles.
Introduction
The 4H-pyran moiety forms the core fragment of numerous biologically active compounds and their importance has been investigated comprehensively. These compounds are known to exhibit diuretic and analgesic,1 anticancer,2 antimicrobial,3 antitumor,4 antiviral,5 anti-proliferative6 and different central nervous system (CNS) activities.7 In addition, they have also been reported to be effective in the treatment of down's syndrome, schizophrenia, and myoclonus diseases.8Apart from their pharmaceutical importance, the 2-amino-3-cyano-4H-pyran derivatives have found applications in cosmetics and pigments9 and as photoactive materials.10 Numerous methodologies have been reported for the synthesis of 4H-benzo[b]pyrans via three-component condensation of aryl aldehydes, dimedone, and malononitrile under various catalytic conditions.11–17 However, with the increasing demand for a cleaner environment and sustainable future, chemists are compelled to design and develop synthetic processes that are more environment-benign. Many alternative green approaches have been explored viz organo catalysts,18–20 ionic liquids21–24 and nanoparticles.25–28 Lately, coated magnetic nanoparticles29–32 have emerged as interesting catalysts due to several advantages they offer. However, despite their promising catalytic properties, only a few of them have been subjected to synthetic applications.
The application of ultrasound in organic syntheses has influenced the rate of reaction as well as the selectivity of the products. The formation of C–C and C–N bonds have been reported to be enhanced by sonication;33 this has been attributed to either the production of new reacting species or due to the increased mechanical effects in the reaction mixtures.34
Thiamine hydrochloride or vitamin B1 (VB1) is a water soluble vitamin containing a pyrimidine and a thiazole ring linked by a methylene bridge. Its catalytic utility has been studied in various organic transformations;35–38 however, its high water solubility posed as a deterrent in terms of reusability. In continuation with our interest in nanoparticle catalyzed organic transformations,39 we explored the possibility of using VB1 as an organocatalyst crafted on silica supported ferrite nanoparticle (Fe2O3@SiO2@VB1) for the synthesis of 4H-benzo[b]pyrans.
Results and discussion
A three-component reaction comprising of aryl aldehydes, malononitrile and dimedone/1,3-cyclohexanedione/4-hydroxycoumarin was carried out in ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) medium. The reaction was facilitated by ultrasound in the presence of Fe2O3@SiO2@VB1 NPs (Scheme 1). The reaction was also carried out with ethyl cyanoacetate instead of malononitrile. In this case, it was found that a higher temperature condition and a longer reaction time were required for a satisfactory yield of the products. On completion of the reaction, the catalyst was easily recovered by using an external magnet.
:4) medium. The reaction was facilitated by ultrasound in the presence of Fe2O3@SiO2@VB1 NPs (Scheme 1). The reaction was also carried out with ethyl cyanoacetate instead of malononitrile. In this case, it was found that a higher temperature condition and a longer reaction time were required for a satisfactory yield of the products. On completion of the reaction, the catalyst was easily recovered by using an external magnet.
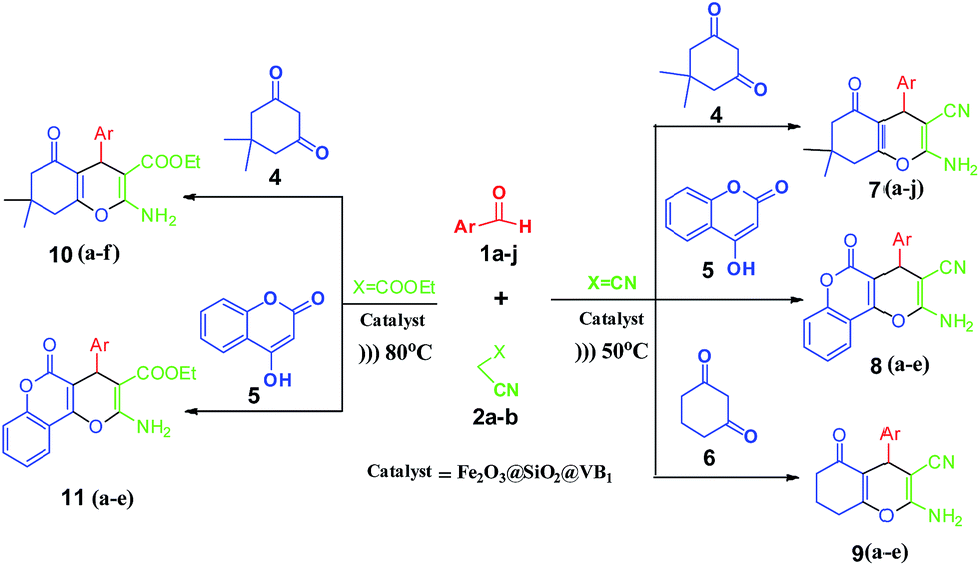 | ||
| Scheme 1 Preparation of benzo[b]pyrans using Fe2O3@SiO2@VB1 NPs as catalyst under ultrasonic condition. | ||
The Fe2O3@SiO2@VB1 NPs were prepared as reported40 by chemical reduction of Fe(II) and Fe(III) salts. SiO2 was then immobilized on the Fe2O3 NPs followed by VB1 functionalization. The synthesized catalyst was then characterized by FT-IR, TEM, EDS and powder XRD.
The FT-IR spectrum (Fig. 1) of Fe2O3@SiO2 shows a band at 800, 1063 and 3435 cm−1 corresponding to the O–Si–O symmetric, asymmetric and –OH stretching respectively. However, the intensity of the –OH peak diminished in the case of VB1 functionalized Fe2O3@SiO2 NP. This can be attributed to the formation of a bond between the Fe2O3@SiO2 NP and VB1 moiety.
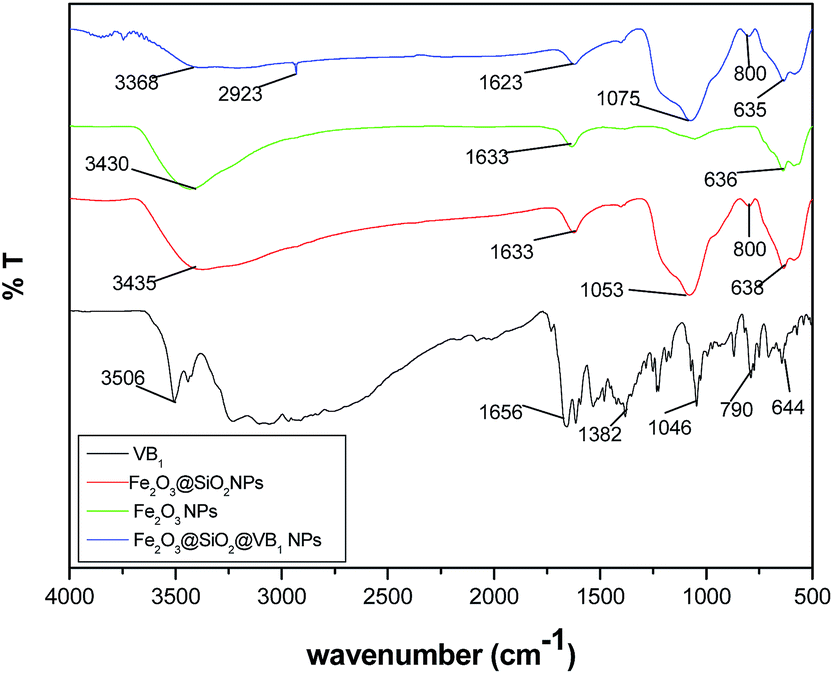 | ||
| Fig. 1 FT-infrared spectra. | ||
The size and structure of the prepared Fe2O3@SiO2@VB1 were evaluated by transmission electron microscopy (TEM) (Fig. 2). The TEM image clearly shows the core–shell structure of the particles and confirms the presence of a coated layer.
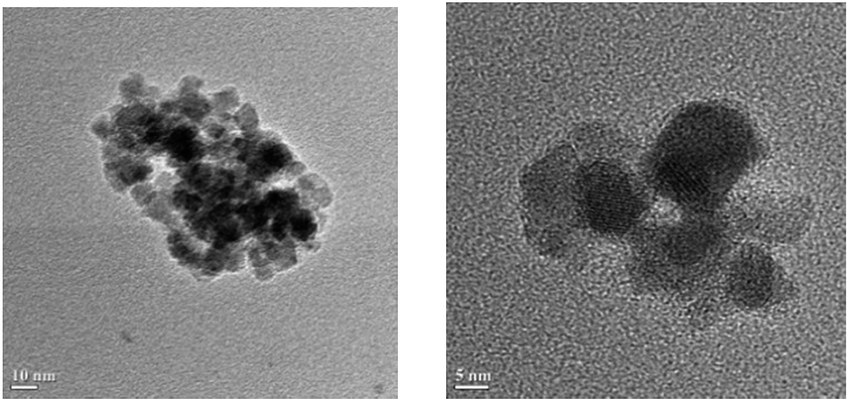 | ||
| Fig. 2 TEM image of Fe2O3@SiO2@VB1 NPs. | ||
The EDS data (Fig. 3) of the prepared catalyst shows the presence of Fe, Si, Cl, N and S peaks, thus confirming the encapsulation of thiamine hydrochloride on the SiO2 coated Fe2O3 nanoparticles.
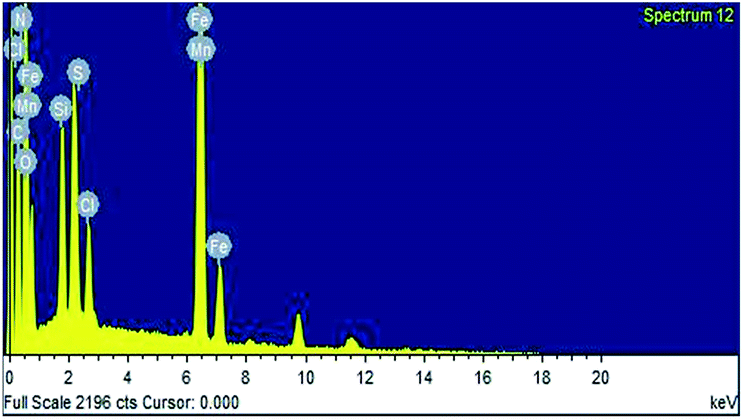 | ||
| Fig. 3 EDS image of Fe2O3@SiO2@VB1 NPs. | ||
The powder XRD plot (Fig. 4) shows diffraction peaks at 2θ = 30.6°, 35.5°, 43.0°, 54.1°, 58.1°, 63.2° corresponding to the (220), (311), (400), (422), (511) and (440) planes.
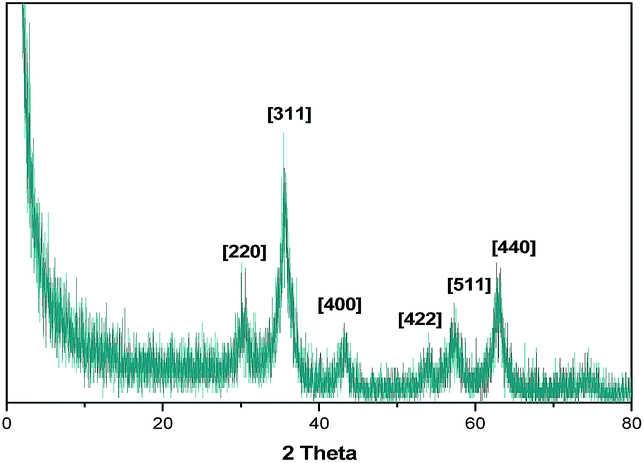 | ||
| Fig. 4 Powder XRD pattern of Fe2O3@SiO2@VB1 NPs. | ||
The efficacy of the catalyst in different reaction media for a model reaction [4-bromobenzaldehyde (1.5 mmol), malononitrile (1.5 mmol) and dimedone (1.5 mmol)] was studied and the observations are represented in Fig. 5. It was observed that the reaction proceeded sluggishly in the case of nonpolar (toluene) and polar aprotic solvents (DMF, DMSO, THF). However, in the case of polar protic solvents like ethanol or water, the resultant yield was >65%. The best results were obtained when ethanol–water (1:4) mixture was used.
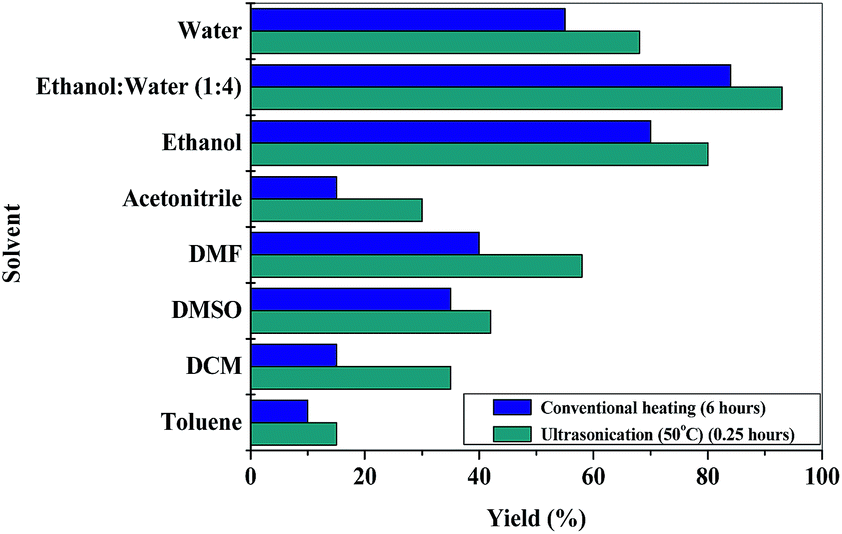 | ||
| Fig. 5 Optimization of the solvent for the preparation of 7b. | ||
The same reaction was studied under both ultrasound irradiation as well as conventional heating method and the time of completion of the reaction was found to be 15 minutes and 6 hours respectively for the formation of 7b. The yield of the product was also found to be much higher in the case of ultrasonication. The plausible mechanistic pathway of the reaction is as given in Scheme 2.
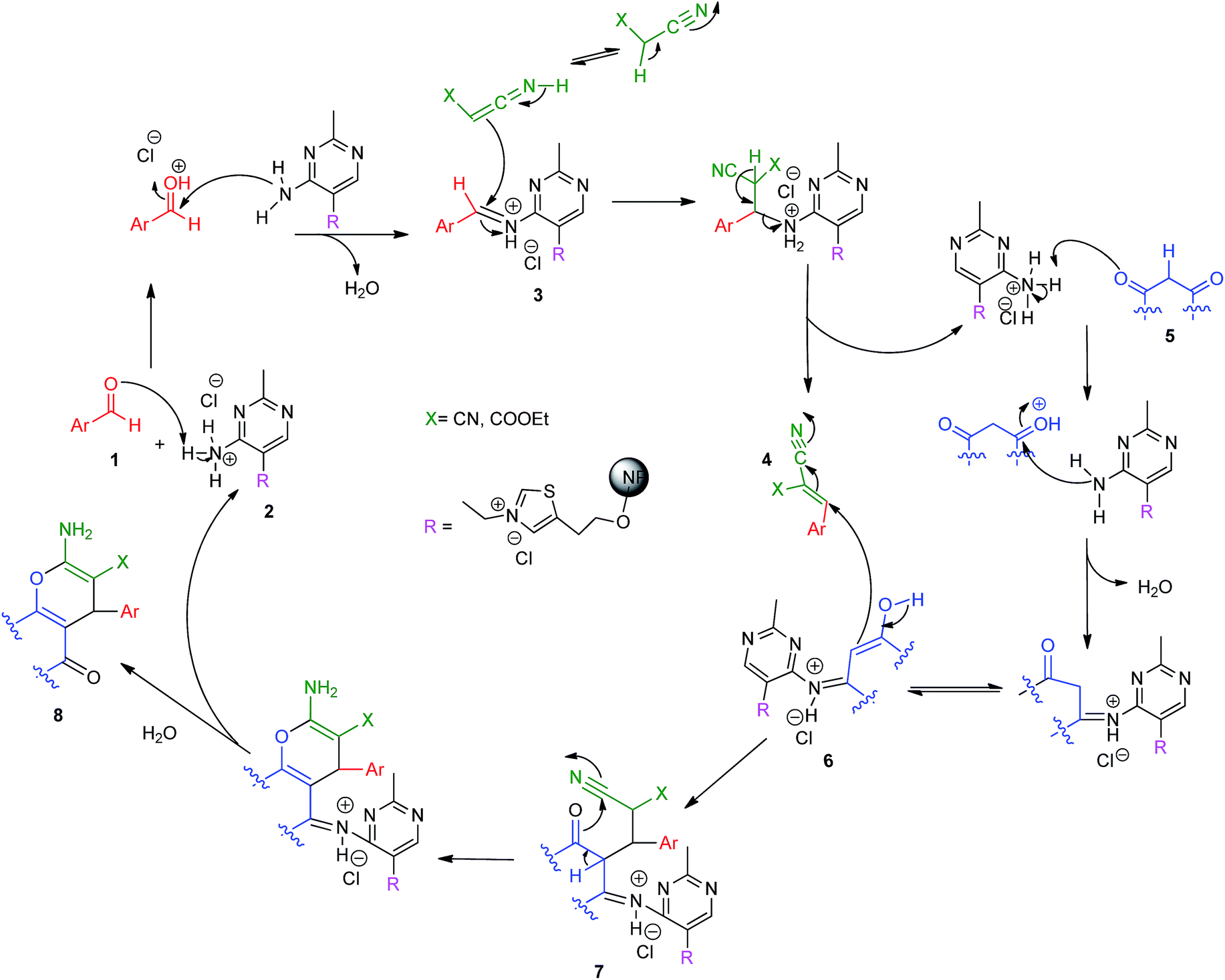 | ||
| Scheme 2 Proposed mechanistic pathway for the formation of benzo[b]pyran derivatives. | ||
Studies were also carried out to optimize the amount of catalyst necessary to promote the model reaction effectively and 8 mg was found to be the ideal amount (Fig. 6).
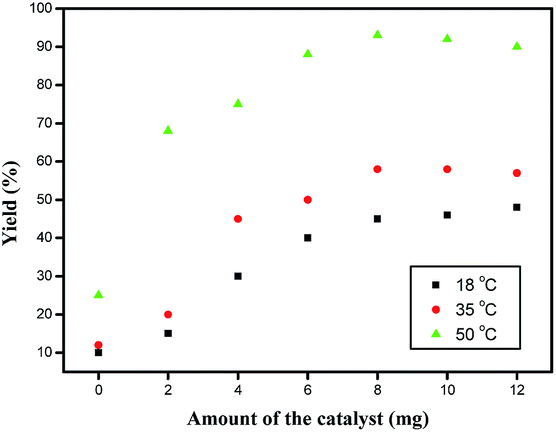 | ||
| Fig. 6 Optimization of the amount of catalyst for the preparation of 7b. | ||
The recyclability of the catalyst was investigated for a model reaction (synthesis of 7b via Scheme 1) and the yields corresponding to six consecutive runs are shown in Fig. 7. The slight decrease in the yield with the number of runs may be attributed to leaching of the catalyst.
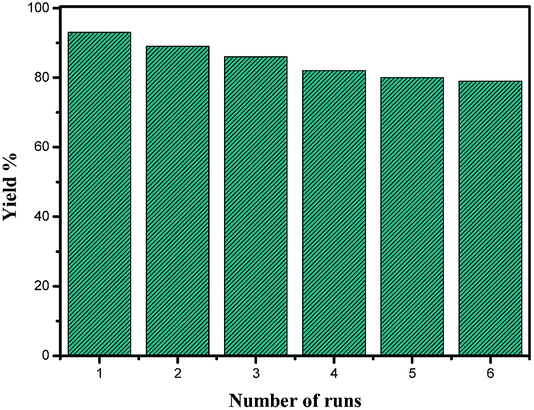 | ||
| Fig. 7 Catalyst reusability plot for the preparation of 7b. | ||
The reaction between ethyl cyanoacetate, malononitrile and an aryl aldehyde (tolualdehyde) can lead to two different products, due to the presence of two electrophilic centres in intermediate IS1; however, it was experimentally found that only one kind of product (A) was formed. Literature survey11–28 of similar reactions also revealed that product A was always formed exclusively irrespective of any catalyst used. In order to investigate the above observation from a theoretical perspective, a Density Functional Theory (DFT) study using B3LYP/6-31+G(d,p) (Fig. 8) was carried out.
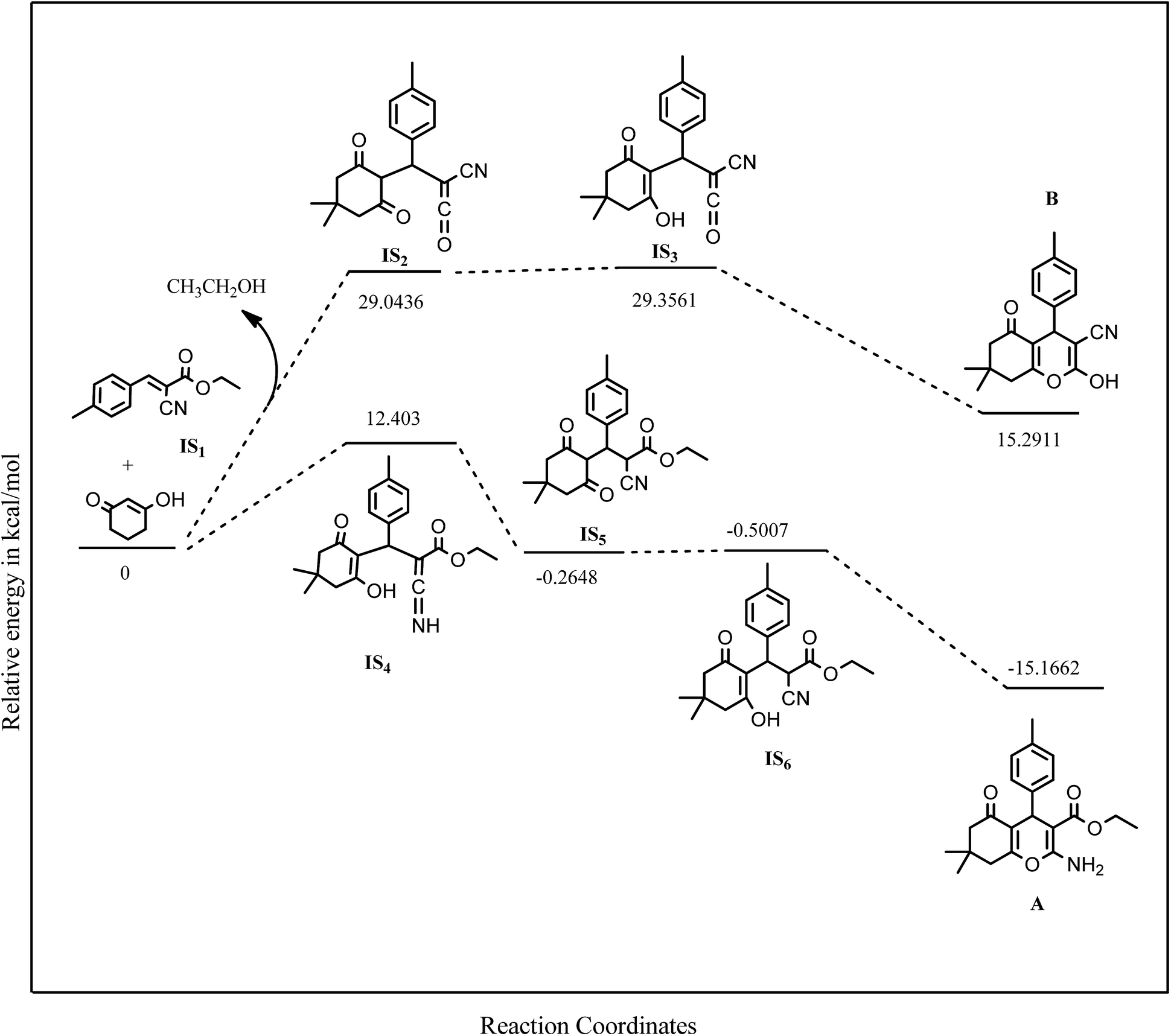 | ||
| Fig. 8 Energy profile calculated for verifying the exclusive formation of (A) on reacting the intermediate (IS1) with dimedone. Relative electronic energies (ΔE) inclusive of zero point energy obtained from B3LYP/6-31+G(d,p). Calculations are given in kcal mol−1. | ||
The geometrical optimizations of all the intermediates were performed using Becke's three-parameter exchange functional41 and the nonlocal correlation functional of Lee, Yang, and Parr42 (B3LYP) in conjunction with the 6-31+G(d,p) basis set.
The energy profile of the reaction pathway leading to products A and B were calculated and the relative electronic energies of the intermediates which were formed by reacting IS1 with dimedone were compared. It was evident from the energy values that the pathway leading to formation of B was not energetically favorable; however, the second pathway leading to formation of A was more energetically favorable. Normally, the transition state energy profiles are important in such kind of mechanistic/pathway studies but, in this case the energy values of the intermediates and products were sufficient to substantiate the assertion.
Single crystal XRD
The single crystal X-ray diffraction (XRD) data were collected at 293 K with Mo Kα radiation (l = 0.71073 Å) using an Agilent Xcalibur (Eos, Gemini) diffractometer equipped with a graphite monochromator. The structures were solved by direct methods and refined by Olex2 refine (Fig. 9).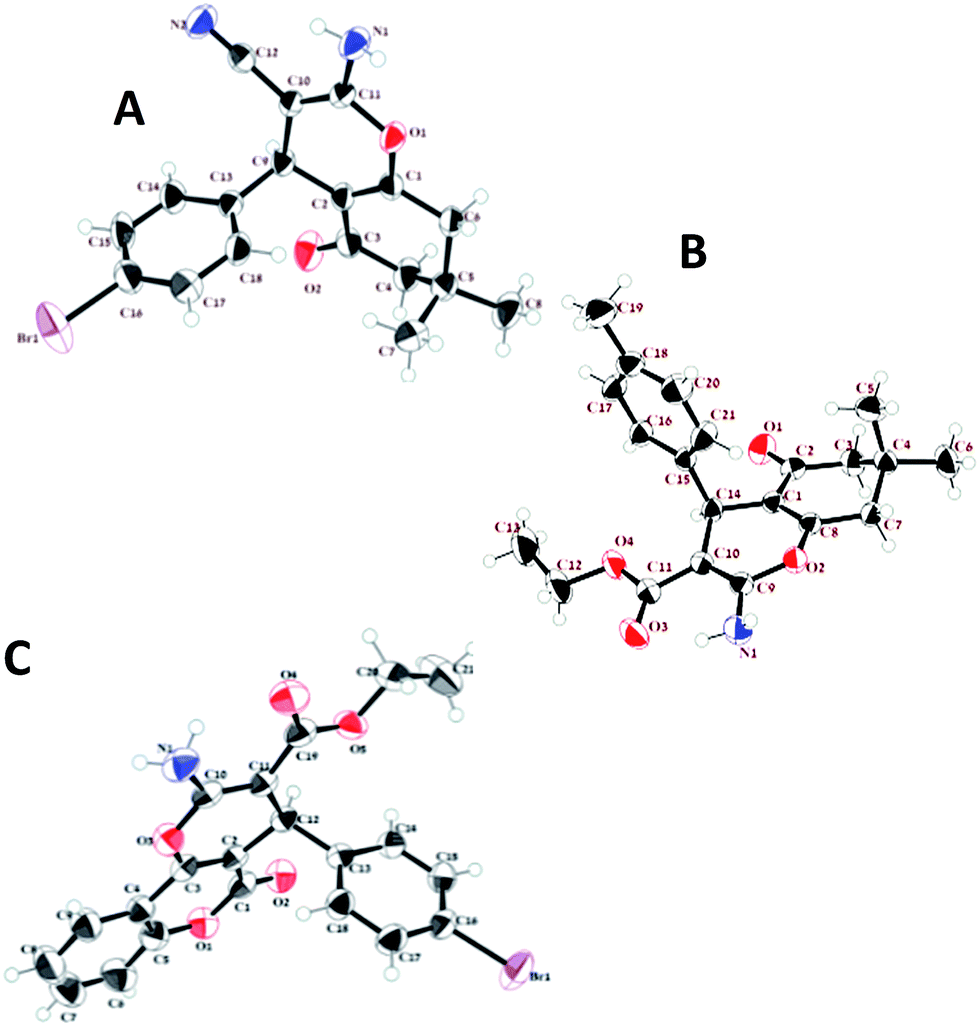
Biological studies
The antimicrobial and antifungal activities of benzopyrans have been documented.5 However, its use as an antidote to combat the toxic effects of pesticides on vertebrates has not been studied till date. A minor study was carried out using 2-amino-7,7-dimethyl-5-oxo-4-(p-tolyl)-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (7f) to investigate its anti-toxic effect against chlorpyrifos, an insecticide, on the morphology of the external gills of Duttaphrynus melanostictus tadpoles. Chlorpyrifos [O,O-diethyl-O-(3,5,6-trichloro-2-pyridyl) phosphorothioate], a broad spectrum organophosphate is the second largest selling43 insecticide in India which is used to control agricultural and household pests.44The study revealed that, exposing the tadpoles to 1 ppm of the insecticide for 48 hours resulted in the bending of the secondary gill filament (respiratory organ) (Fig. 10A). However, when the tadpoles were treated with a solution of 1 ppm of the insecticide mixed with 0.25 ml of the sample (0.05 moles of 7f in 5 ml acetone) in 150 ml dechlorinated water, it was observed that there was no bending of the gills (Fig. 10D) and the morphology was similar to that of the controlled condition (Fig. 10B). On exposure of the tadpoles to the solution of the sample only, no distinctive change in the morphology of the gills was observed (Fig. 10C). Thus it can be inferred that compound (7b) suppress the toxic effect of the insecticide to a large extent.
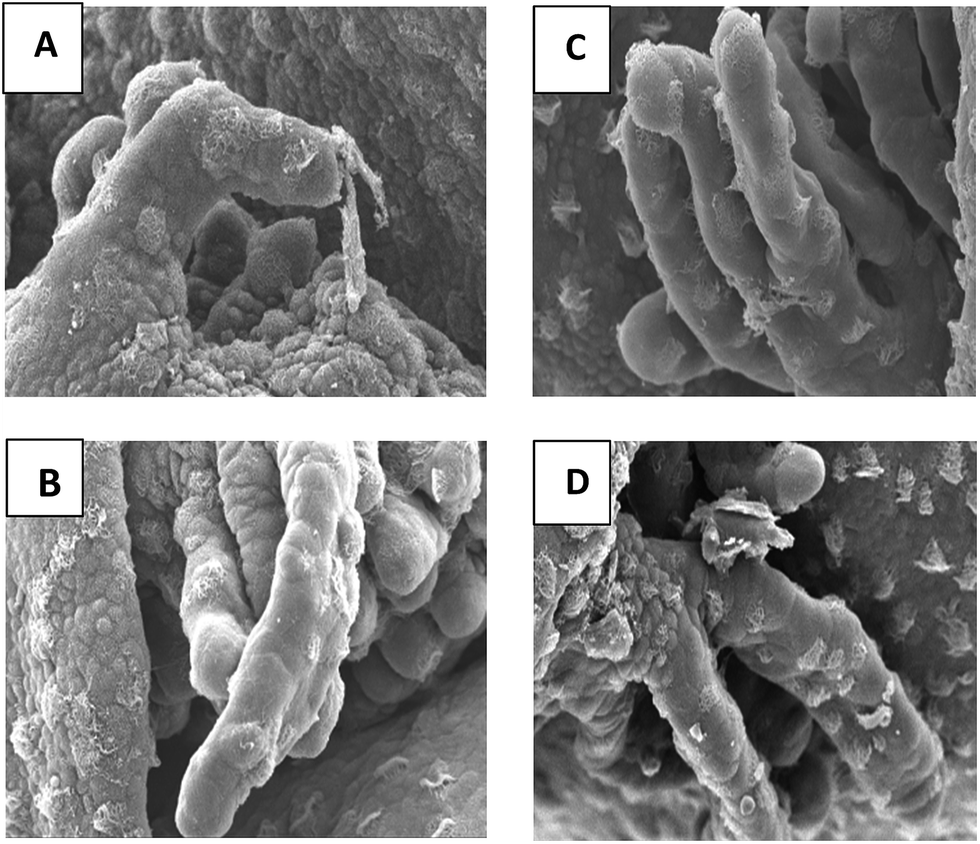 | ||
| Fig. 10 Effect of chlorpyrifos on the external gills of Duttaphrynus melanostictus tadpole (Gosner stage 23) in: (A) 1 ppm of chlorpyrifos after 48 hours of treatment. (B) Controlled condition (C) 0.25 ml of sample (D) 1 ppm of chlorpyrifos mixed with 0.25 ml of sample. | ||
Conclusion
In summary, we have demonstrated an efficient one-pot three-component synthesis of benzo[b]pyran derivatives using a novel and magnetically retrievable Fe2O3@SiO2@VB1 nano-organocatalyst. It was also found that application of ultrasound aided in reducing the reaction time and improving the product yield, thus exhibiting the superiority of the ultrasonication protocol. Considering the ease of catalyst recovery and reusability combined with mild reaction conditions; this protocol can be further exploited for various synthetic approaches related to the formation of C–C, C–O bonds, etc. In addition, the biological importance of 2-amino-7,7-dimethyl-5-oxo-4-(p-tolyl)-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile in suppressing the toxic effect of chlorpyrifos (an insecticide) on vertebrates have been investigated and this can be further extended to in vitro studies in the near future.Experimental section
General information
All the chemicals involved in the synthesis were purchased from Merck, Sigma Aldrich and Alfa Aesar and were used without further purification. The ultra-sonication reaction was carried out in an ultrasonic bath of make Bandelin Sonorex Model RK 52H Incarp Germany (50/60 Hz, 60/240 W). The purity of the synthesized products was confirmed by melting point, infrared (FT-IR), 1H-NMR, 13C-NMR and mass spectrometry. The melting points were determined in open capillaries in an Optics Technology melting point apparatus and are uncorrected. FT-IR spectra were recorded with a Perkin Elmer Spectrum BX FT-IR apparatus (λmax in cm−1) on KBr disks. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance II-400 spectrometer in CDCl3 and DMSO-d6 (chemical shifts in δ with TMS as the internal standard). Mass spectra were recorded on WATERS (ZQ-4000) mass spectrometer. Single crystal XRD data were obtained with an Xcalibur-Eos-Gemini instrument and powder XRD analysis was conducted with an X'Pert Pro instrument. The TEM images were captured using a transmission electron microscope of JEM-2100 make, 200 kV (JEOL) and SEM and EDS imaging were carried out with scanning electron microscope of JSM-6360 (JEOL) make. All reactions were monitored by thin-layer chromatography (TLC) using pre-coated aluminum sheets (silica gel 60 F 254 0.2 mm thickness).:1), 8 mg of Fe2O3@SiO2@VB1 NPs was added. The solution was then subjected to ultrasonic irradiation for the time indicated in Table 1 at 50/80 °C. The progress of the reaction was monitored by TLC and on completion, the catalyst was recovered magnetically. The mixture was then poured into ice cold water where the solid separates out. The product was filtered and then recrystallized from warm ethanol.
| Entry | X | 1,3-dicarbonyl | Ar | Product | Ultrasound irradiation | Conventional heating | ||
|---|---|---|---|---|---|---|---|---|
| Time (min) | Yielda (%) | Time (h) | Yielda (%) | |||||
| a Isolated yield. | ||||||||
| 1 | CN | 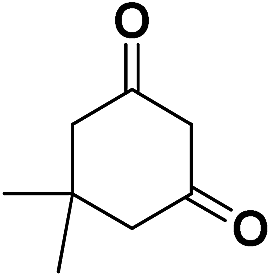 |
C6H5 | 7a | 15 | 90 | 6 | 84 |
| 2 | CN | 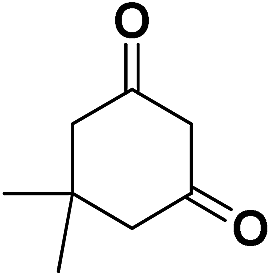 |
4-BrC6H4 | 7b | 15 | 93 | 6 | 86 |
| 3 | CN | 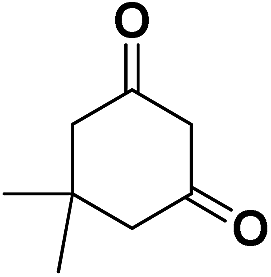 |
4-ClC6H4 | 7c | 15 | 91 | 6 | 80 |
| 4 | CN | 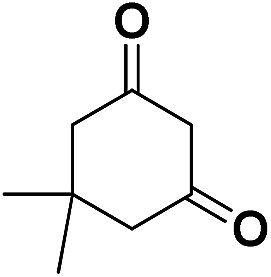 |
4-OCH3C6H4 | 7d | 18 | 88 | 6 | 64 |
| 5 | CN |  |
4-OHC6H4 | 7e | 15 | 92 | 6 | 75 |
| 6 | CN |  |
4-CH3C6H4 | 7f | 15 | 93 | 6 | 85 |
| 7 | CN |  |
2-NO2C6H4 | 7g | 20 | 88 | 6 | 70 |
| 8 | CN |  |
2-ClC6H4 | 7h | 18 | 91 | 6 | 78 |
| 9 | CN | 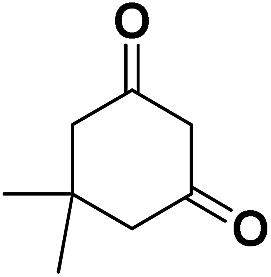 |
3-OCH3C6H4 | 7i | 20 | 89 | 6 | 71 |
| 10 | CN | 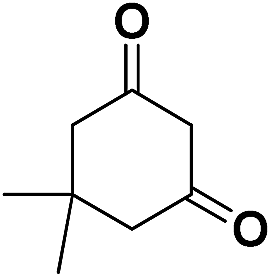 |
C6H5CHCH | 7j | 18 | 88 | 6 | 65 |
| 11 | CN | 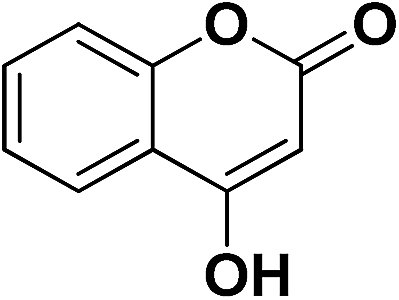 |
C6H5 | 8a | 25 | 90 | 6 | 80 |
| 12 | CN | 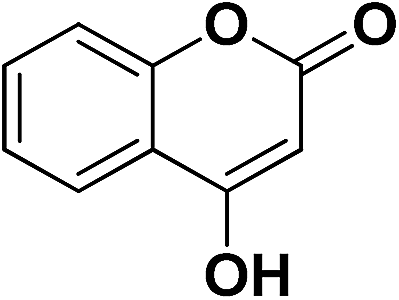 |
4-CH3C6H4 | 8b | 25 | 88 | 6 | 72 |
| 13 | CN | 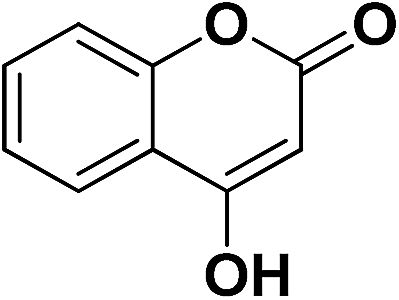 |
4-ClC6H4 | 8c | 30 | 90 | 6 | 76 |
| 14 | CN | 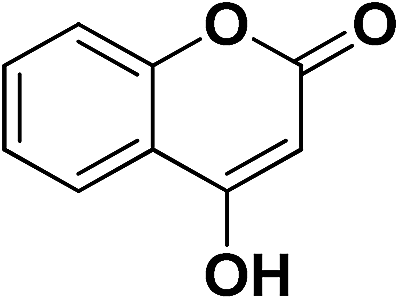 |
4-OHC6H4 | 8d | 25 | 88 | 6 | 65 |
| 15 | CN | 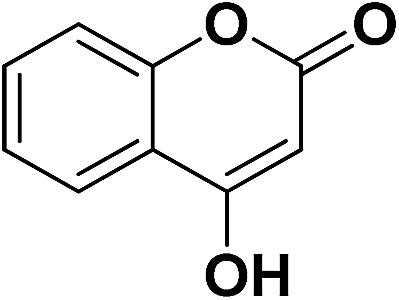 |
C6H5CHCH | 8e | 25 | 85 | 6 | 65 |
| 16 | CN | 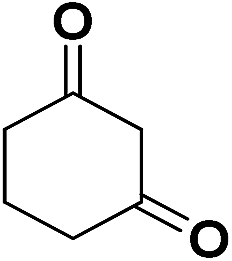 |
C6H5 | 9a | 20 | 90 | 6 | 66 |
| 17 | CN | 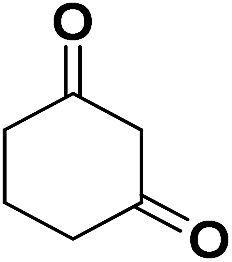 |
4-CH3C6H4 | 9b | 20 | 92 | 6 | 70 |
| 18 | CN | 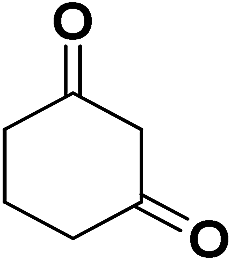 |
4-NO2C6H4 | 9c | 20 | 88 | 6 | 65 |
| 19 | CN | 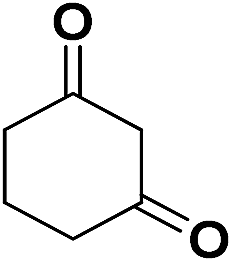 |
2-ClC6H4 | 9d | 25 | 90 | 6 | 68 |
| 20 | CN | 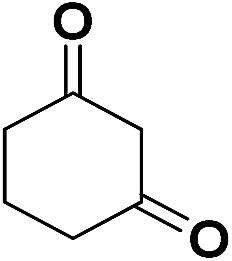 |
4-OHC6H4 | 9e | 15 | 90 | 6 | 73 |
| 21 | CO2Et |  |
4-CH3C6H4 | 10a | 30 | 82 | 6 | 68 |
| 22 | CO2Et |  |
4-OCH3C6H4 | 10b | 35 | 80 | 6 | 65 |
| 23 | CO2Et |  |
4-ClC6H4 | 10c | 30 | 80 | 6 | 65 |
| 24 | CO2Et |  |
4-BrC6H4 | 10d | 35 | 80 | 6 | 68 |
| 25 | CO2Et | 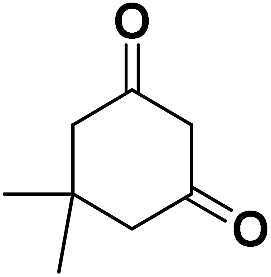 |
2-ClC6H4 | 10e | 35 | 78 | 6 | 65 |
| 25 | CO2Et | 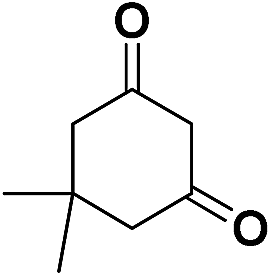 |
3-OCH3C6H4 | 10f | 35 | 78 | 6 | 60 |
| 26 | CO2Et | 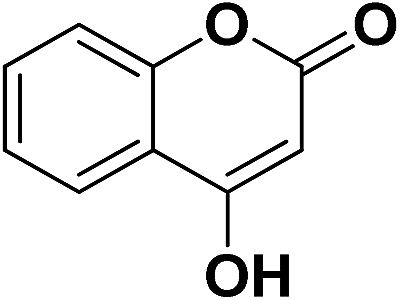 |
4-CH3C6H4 | 11a | 40 | 75 | 6 | 58 |
| 27 | CO2Et | 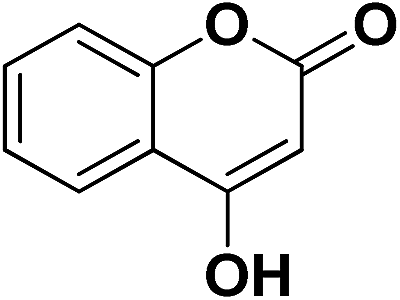 |
4-OCH3C6H4 | 11b | 40 | 72 | 6 | 55 |
| 28 | CO2Et |  |
4-ClC6H4 | 11c | 40 | 75 | 6 | 58 |
| 30 | CO2Et |  |
2-ClC6H4 | 11e | 45 | 70 | 6 | 55 |
Selected spectral data
Acknowledgements
R. Nongrum would like to thank Sankardev College, Shillong and UGC for the award of teacher fellowship under the Faculty Development Programme. The authors are also grateful to Ms Makroni Lily for her valuable help and guidance. The analytical assistance from the Department of Chemistry-NEHU, SAIF–NEHU, Param Shreesh Supercomputer-NEHU, IIT-Guwahati is acknowledged.References
- L. Bonsignore, G. Loy, D. Secci and A. Calignano, Eur. J. Med. Chem., 1993, 28, 517–520 CrossRef CAS.
- W. Kemnitzer, S. Kasibhatla, S. Jiang, H. Zhang, J. Zhao, S. Jia, L. Xu, C. Crogan-Grundy, R. Denis, N. Barriault, L. Vaillancourt, S. Charron, J. Dodd, G. Attardo, D. Labrecque, S. Lamothe, H. Gourdeau, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2005, 15, 4745–4751 CrossRef CAS PubMed.
- M. M. Khafagy, A. H. Abd El-Wahab, F. A. Eid and A. M. El-Agrody, Il Farmaco, 2002, 57, 715–722 CrossRef CAS PubMed.
- D. Kessel, J. G. Belton and D. K. J. G. Belton, Cancer Res., 1975, 35, 3735–3740 CAS.
- P. W. Smith, S. L. Sollis, P. D. Howes, P. C. Cherry, I. D. Starkey, K. N. Cobley, H. Weston, J. Scicinski, A. Merritt, A. Whittington, P. Wyatt, N. Taylor, D. Green, R. Bethell, S. Madar, R. J. Fenton, P. J. Morley, T. Pateman and A. Beresford, J. Med. Chem., 1998, 41, 787–797 CrossRef CAS PubMed.
- C. P. Dell and C. W. Smith, EP 0537949 B1, 1998.
- F. Eiden and F. Denk, Arch. Pharm., 1991, 324, 353–354 CrossRef CAS PubMed.
- C. S. Konkoy, D. B. Fick, S. X. Cai, N. C. Lan and J. F. W. Keana, Int. Appl. WO, 75 123, 2000Chem. Abstr., 2001, 134, 29313a.
- G. P. Ellis, The Chemistry of Heterocyclic Compounds: Chromenes, Chromanes, and Chromones, ed. A. Weissberger and E. C. Taylor, Wiley, New York, 1977, vol. 31, p. 13 Search PubMed.
- D. Armesto, W. M. Horspool, N. Martin, A. Ramos and C. Seoane, J. Org. Chem., 1989, 54, 3069–3072 CrossRef CAS.
- R. A. Mekheimer, A. M. Abdelhameed, S. M. Mohamed and K. U. Sadek, Green Chem. Lett. Rev., 2010, 3, 161–163 CrossRef CAS.
- G. Sabitha, K. Arundhathi, K. Sudhakar, B. S. Sastry and J. S. Yadav, Synth. Commun., 2009, 39, 433–442 CrossRef CAS.
- R. Hekmatshoar, S. Majedi and K. Bakhtiari, Catal. Commun., 2008, 9, 307–310 CrossRef CAS.
- S. Balalaie, M. Bararjanian, M. Sheikh-Ahmadi, S. Hekmat and P. Salehi, Synth. Commun., 2007, 37, 1097–1108 CrossRef CAS.
- R. S. Bhosale, C. V. Magar, K. S. Solanke, S. B. Mane, S. S. Choudhary and R. P. Pawar, Synth. Commun., 2007, 37, 4353–4357 CrossRef CAS.
- G. Ziarani, A. Abbasi, A. Badiei and Z. Aslani, E-J. Chem., 2011, 8, 293 CrossRef CAS.
- J. Li, W. Xu, L. Yang and T. Li, Synth. Commun., 2004, 34, 4565–4571 CrossRef CAS.
- N. M. H. Elnagdi and N. S. Al-Hokbany, Molecules, 2012, 17, 4300–4312 CrossRef CAS PubMed.
- S. Balalaie, M. Bararjanian, A. M. Amani and B. Movassagh, Synlett, 2006, 263, 263–266 CrossRef.
- U. R. Pratap, D. V. Jawale, P. D. Netankar and R. A. Mane, Tetrahedron Lett., 2011, 52, 5817–5819 CrossRef CAS.
- S. Gao, C. H. Tsai, C. Tseng and C. Yao, Tetrahedron, 2008, 64, 9143–9149 CrossRef CAS.
- S. Balalaie, M. Sheikh-Ahmadi and M. Bararjanian, Catal. Commun., 2007, 8, 1724–1728 CrossRef CAS.
- S. Gurumurthi, V. Sundari and R. Valliappan, E-J. Chem., 2009, 6, S466–S472 CrossRef CAS.
- D. Shi, J. Mou, Q. Zhuang and X. Wang, J. Chem. Res., 2004, 821–823 CrossRef CAS.
- M. M. Heravi, S. Yahya Shirazi Beheshtiha, M. Dehghani and N. Hosseintash, J. Iran. Chem. Soc., 2015, 12, 2075–2081 CrossRef CAS.
- A. Maleki, M. Ghassemi and V. Eskandarpour, in Proceedings of the 18th International Electronic Conference on Synthetic Organic Chemistry, MDPI, Basel, Switzerland, 2014, vol. 462, p. a037 Search PubMed.
- L. Edjlali and R. H. Khanamiri, Monatshefte für Chemie -Chemical Monthly, 2016, 147, 1221–1225 CrossRef CAS.
- S. Banerjee and A. Saha, New J. Chem., 2013, 37, 4170 RSC.
- S. Luo, X. Zheng, H. Xu, X. Mi, L. Zhang and J. P. Cheng, Adv. Synth. Catal., 2007, 349, 2431–2434 CrossRef CAS.
- H. R. Shaterian and M. Aghakhanizadeh, Catal. Sci. Technol., 2013, 3, 425–428 CAS.
- S. M. Sadeghzadeh and M. A. Nasseri, Catal. Today, 2013, 217, 80–85 CrossRef CAS.
- E. Karaoğlu, A. Baykal, M. Şenel, H. Sözeri and M. S. Toprak, Mater. Res. Bull., 2012, 47, 2480–2486 CrossRef.
- D. H. Bremner, Ultrason. Sonochem., 1994, 1, S119–S124 CrossRef CAS.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2006, 35, 180–196 RSC.
- M. Lei, L. Ma and L. Hu, Tetrahedron Lett., 2009, 50, 6393–6397 CrossRef CAS.
- O. B. Pawar, F. R. Chavan, V. S. Suryawanshi, V. S. Shinde and N. D. Shinde, J. Chem. Sci., 2013, 125, 159–163 CrossRef CAS.
- M. Lei, L. Ma and L. Hu, Synth. Commun., 2011, 41, 1969–1976 CrossRef CAS.
- I. R. Siddiqui, P. Rai, Rahila, A. Srivastava, A. Srivastava and A. Srivastava, New J. Chem., 2013, 37, 3798 RSC.
- N. G. Singh, R. Nagarajaprakash, J. W. S. Rani, C. Kathing, R. Nongrum and R. Nongkhlaw, New J. Chem., 2015, 39, 3908–3915 RSC.
- N. G. Singh, M. Lily, S. P. Devi, N. Rahman, A. Ahmed, A. K. Chandra and R. Nongkhlaw, Green Chem., 2016, 18, 4216–4227 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. Maharajan, Y. Narayanasamy, V. Ganapiriya and K. Shanmugavel, J. Basic Appl. Zool., 2015, 72, 104–112 CrossRef CAS.
- N. Deb and S. Das, Curr. World Environ., 2013, 8, 77–84 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1477443, 1477627 and 1477628. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra24108e |
| This journal is © The Royal Society of Chemistry 2016 |