
Microarrays for the study of compartmentalized microorganisms in alginate microbeads and (W/O/W) double emulsions†
Abstract
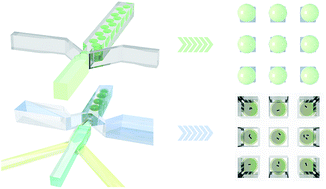
Understanding of cells has mainly been achieved through analysis of average properties of large cellular populations. The increased awareness of cellular heterogeneity in various biological systems, spans from single bacterial cells to human tissue calls for alternative strategies. The need to understand cellular heterogeneity has motivated the development of instrumentation, protocols, and methods for analyzing single cells. In this paper we demonstrate how microorganisms encapsulated in micron sized alginate microbeads with droplet based microfluidics can be immobilized on microarrays on glass surfaces by virtue of micro contact printing. The microarray presentation opens for efficient microscopic inspection of high numbers of single cells as well as colony picking by selective microbead aspiration as demonstrated herein. We also present a double emulsion (W/O/W) micro-array arrangement strategy for the investigation of swimming microorganisms with a focus on the widely studied P. putida.
 Please wait while we load your content...
Please wait while we load your content...

