
Analysis of burn-in photo degradation in low bandgap polymer PTB7 using photothermal deflection spectroscopy†
Abstract
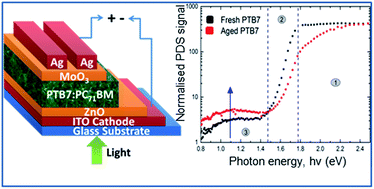
The efficiency of organic photovoltaic devices continues to increase; however, their limited stability is currently a barrier to the commercial prospects of the technology. Burn-in photo degradation, caused by continuous illumination under a light source, can cause a significant reduction in device performance. Our aim was to investigate this degradation pathway for the high-efficiency polymer PTB7, which was compared to the well-studied P3HT:PC71BM material system. In this study, we compared the burn-in aging profile for organic solar cells containing either P3HT or PTB7 as the donor polymer. This showed that PTB7:PC71BM solar cells exhibit a severe initial reduction in performance, due mainly to reduced short circuit current density (Jsc), during the 5 hour test period. P3HT:PC71BM cells were relatively stable during this test. Photothermal deflection spectroscopy (PDS), which provides sensitive measurement of sub bandgap absorption, was employed to discover the underlying mechanism causing this discrepancy. In PTB7-based devices, a significant increase in sub bandgap absorption was observed after illumination, which was attributed to the formation of sub bandgap trap states. This mechanism was identified as a contributing factor to the severe burn-in for PTB7-based organic solar cells. No such increase was observed for P3HT:PC71BM films.
 Please wait while we load your content...
Please wait while we load your content...

