
Facile synthesis of a dimeric titanium(IV) complex with terminal Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O moieties and its application as a catalyst for the cycloaddition reaction of CO2 to epoxides†
O moieties and its application as a catalyst for the cycloaddition reaction of CO2 to epoxides†
Hyejin Kim‡
a,
Sung Ho Choi‡a,
Duseong Ahna,
Yoseph Kima,
Ji Yeon Ryu b,
Junseong Lee*b and
Youngjo Kim*a
b,
Junseong Lee*b and
Youngjo Kim*a
aDepartment of Chemistry, BK21+ Program Research Team, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea. E-mail: ykim@chungbuk.ac.kr; Fax: +82 43 267 2279; Tel: +82 43 261 3395
bDepartment of Chemistry, Chonnam National University, Gwangju 61186, Republic of Korea. E-mail: leespy@chonnam.ac.kr; Fax: +82 62 530 3389; Tel: +82 62 530 3371
First published on 7th October 2016
Abstract
In this report, facile and exclusive synthesis of dimeric titanium(IV) complex with a terminal Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety from the reaction between novel pyridine-based tridentate ligand (LH2) and Ti(O-i-Pr)4 under the bubbling of wet air is presented. Alternatively, the same dimeric Ti complex was obtained via wet air bubbling of monomeric LTi(O-i-Pr)2 or addition of the same equiv. of H2O into LTi(O-i-Pr)2. All compounds including LH2 and two titanium complexes were characterized by single crystal X-ray analyses. Newly synthesized terminal oxo-titanium compound is the first example of structurally characterized dimeric terminal oxo-titanium compound having no TiO→Ti bonds. Two titanium complexes were used as effective catalysts for the cycloaddition of CO2 to propylene oxide in the presence of various kinds of cocatalysts such as n-Bu4PBr, n-Bu4NI, n-Bu4NBr, n-Bu4NCl, PPNCl, and DMAP.
O moiety from the reaction between novel pyridine-based tridentate ligand (LH2) and Ti(O-i-Pr)4 under the bubbling of wet air is presented. Alternatively, the same dimeric Ti complex was obtained via wet air bubbling of monomeric LTi(O-i-Pr)2 or addition of the same equiv. of H2O into LTi(O-i-Pr)2. All compounds including LH2 and two titanium complexes were characterized by single crystal X-ray analyses. Newly synthesized terminal oxo-titanium compound is the first example of structurally characterized dimeric terminal oxo-titanium compound having no TiO→Ti bonds. Two titanium complexes were used as effective catalysts for the cycloaddition of CO2 to propylene oxide in the presence of various kinds of cocatalysts such as n-Bu4PBr, n-Bu4NI, n-Bu4NBr, n-Bu4NCl, PPNCl, and DMAP.
Introduction
The efficient capture and utilization of carbon dioxide, a valuable C1 building block, have been an important theme environmentally and chemically for a long time.1 Thus, the development of chemical methods to activate and convert CO2 into methanol, formic acid, urea, cyclic carbonate, and polycarbonates has drawn significant attention in recent years. Among them, cyclic carbonates from CO2 and epoxides have been produced since the 1950s and find use as aprotic solvents,2 electrolytes for secondary batteries,3 and monomers for polymers4 and pharmaceutical intermediates.5A variety of metal-based catalytic systems, including those with Cr, Fe, Co, Ni, Cu, Al, and so on, have been developed to couple CO2 and epoxides.6–10 Titanium, the seventh most abundant metal in earth's crust (e.g., Al, Fe, Ca, Na, K, Mg, Ti in the order), could be an attractive metal candidate due to its low toxicity and high Lewis acidity.10 Although Ti-based complexes have been widely used as catalysts for various organic transformation and polymer synthesis,11–13 a few examples of Ti-based catalytic systems for the cycloaddition of CO2 to epoxide have been reported in the literature.14–20
Due to high oxophilic property of titanium metal, many titanium compounds could easily react with oxygen-donating sources such as H2O, O2, and CO to form oxotitanium complexes. In general, oxotitanium compounds can be classified as two types of complexes depending on functional connectivity with: (1) quite common and bridging [Ti–O–Ti]6+ unit and (2) very rare and terminal [TiO]2+ moiety. Several structurally characterized titanium complexes with terminal TiO group were reported in the literature.21–33 Interestingly, only titanium compounds chelated by sterically encumbering ligands such as tetradentate porphyrin-type ligands,21–24 two bidentate ketiminate ligands,25 two Cp* ligands,26–28 tetradentate salen-type ligand,29 tridentate Me3tacn ligands (where Me3tacn is 1,4,7-trimethyl-1,4,7-triazacyclononae),30,31 two bidentate benzamidinate ligands,32 and tridentate PNP scaffold ligand33 could generate terminal oxo product. Their major coordination geometries around titanium are square pyramidal.21–25 Others are trigonal planar,26 tetrahedral,27,28 trigonal bipyramidal,29 and octahedral.30–33 Interestingly, structurally characterized terminal oxo-titanium complexes are all monomeric though some dimeric Ti compounds with TiO→Ti bonds are reported in the literature.34–37 In those cases, Ti–O bond distances are about 1.80 Å, which is much longer than pure TiO bond distances of 1.65 Å.21–33 To our best knowledge, real dimeric terminal oxotitanium compound without TiO→Ti bonds has never been reported in the literature. In this regard, we focused on the development of new dimeric or polymeric terminal oxo-Ti compounds having no TiO→Ti bonds.
Whereas a lot of catalytic systems for CO2 conversion into cyclic carbonates have been reported in the literature, the research of Ti-based complexes is an unexplored field.14–17 Previous work in our group explored the importance of electronic effect at Ti center on catalytic activity for cycloaddition reaction.16 During our continuous search for new Ti-based catalysts, we report here the synthesis and characterization of diisopropoxytitanium complex 1 containing the novel 2-tridentate aminopyridine-based ligand LH2 and its facile transformation to terminal oxo complex 2, along with their catalytic behavior in the cycloaddition reaction of CO2 to epoxides.
Experimental
Methods and materials
All manipulations were carried out under an atmosphere of dinitrogen by using standard Schlenk-type glassware on a dual manifold Schlenk line in a glove box.38 Dinitrogen was deoxygenated using activated Cu catalyst and dried with calcium sulfate.39 All chemicals were purchased from Aldrich and used as supplied unless otherwise indicated. All solvents such as toluene, diethyl ether, and n-hexane, were dried by distillation from sodium diphenylketyl under dinitrogen and were stored over 3 Å activated molecular sieves. CDCl3 and CD3OD were dried over 4 Å activated molecular sieves and used after vacuum transfer to a Schlenk tube equipped with a J. Young valve.1H and 13C NMR spectra were recorded at ambient temperature on a 400 MHz NMR spectrometer using standard parameters. All chemical shifts are reported in δ units with reference to the peaks of residual CDCl3 (δ 7.24, 1H NMR; δ 77.0, 13CNMR) or CD3OD (δ 3.30, 1H NMR; δ 49.0, 13C NMR). Elemental analyses and mass data measurements were performed with EA 1110-FISONS analyser and VG Auto Spec., respectively.
Synthesis of HOCMe2CH2NC5H4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) NCH2CMe2OH (LH2)
NCH2CMe2OH (LH2)

Isobutylene oxide (9.01 g, 125 mmol) and 2-aminopyridine (4.71 g, 50.0 mmol) were added to a 20 mL screw cap vial containing stirring bar. The vial was tightly sealed by Teflon tape and paraffin film. The mixture was maintained at room temperature for overnight and was then heated for 3 days at 75 °C. The removed of volatiles at reduced pressure gave the desired product LH2 (11.8 g, 99.0%) as a yellow powder. 1H NMR (CDCl3, 400.15 MHz): δ 7.76 (br, 1H, OH), 7.03 (t, J = 7.9 Hz 1H, Ar–H), 6.97 (d, 1H, J = 6.8 Hz, Ar–H), 6.45 (d, 1H, J = 9.4 Hz, Ar–H), 5.81 (t, 1H, J = 6.6 Hz, Ar–H), 3.97 (s, 2H, ArNCH2–), 2.99 (s, 2H, –NCH2–), 2.44 (br, 1H, OH), 1.27 (s, 6H, –CMe2OH), 1.23 (s, 6H, –CMe2OH). 1H NMR (CD3OD, 400.15 MHz): δ 7.28 (dd, J = 6.8 and 1.2 Hz, 1H, Ar–H), 7.13 (t, J = 7.9 Hz, 1H, Ar–H), 6.56 (d, J = 9.5 Hz, 1H, Ar–H), 5.92 (dt, J = 6.7 and 1.2 Hz, 1H, Ar–H), 4.04 (s, 2H, ArNCH2–), 3.03 (s, 2H, –NCH2–), 1.25 (s, 6H, –CMe2OH), 1.19 (s, 6H, –CMe2OH). 13C NMR (CDCl3, 100.63 MHz): δ 156.7, 139.8, 136.3, 113.2, 102.5 (Ar), 71.30 (ArNCH2–), 70.56 (–NCH2–), 64.00 (–CMe2OH), 58.80 (–CMe2OH), 27.99 (–CMe2OH), 27.61 (–CMe2OH). 13C NMR (CD3OD, 100.63 MHz): δ 157.9, 141.8, 138.0, 113.6, 103.9 (Ar), 72.64 (ArNCH2–), 72.36 (–NCH2–), 64.76 (–CMe2OH), 59.92 (–CMe2OH), 27.95 (–CMe2OH), 27.86 (–CMe2OH). HRMS m/z calcd for C13H22N2O2 239.1760, found 239.1754.
Synthesis of LTi(O-i-Pr)2 (1)

To a stirred yellow solution of LH2 (0.596 g, 2.50 mmol) in 30 mL of toluene was added Ti(O–i–Pr)4 (0.711 g, 2.50 mmol) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for overnight. The residue, obtained by removing the solvent under vacuum, was recrystallized in toluene. The desired product 1 was isolated as yellow crystals after the solution remained at −15 °C in a refrigerator for a few days (94.7%, 0.953 g). 1H NMR (CDCl3, 400.13 MHz): δ 7.18 (m, 1H, Ar–H), 7.15 (dt, J = 1.2 and 5.6 Hz, 1H, Ar–H), 6.61 (d, J = 9 Hz, 1H, Ar–H), 6.10 (td, J = 6.6 and 1.3 Hz, 1H, Ar–H), 4.63 (m, 2H, –OCHMe2), 4.04 (br, 2H, ArNCH2–), 3.28 (br, 2H, –NCH2–), 1.29 (s, 6H, –CMe2O–), 1.23 (s, 6H, –CMe2O–), 1.15 (d, J = 6.1 Hz, 6H, –OCHMe2), 1.11 (d, J = 6.1 Hz, 6H, –OCHMe2). 1H NMR (CD3OD, 400.15 MHz): δ 7.58 (s, 1H, Ar–H), 7.49 (d, J = 4.9 Hz, 1H, Ar–H), 6.90 (d, J = 5.4 Hz, 1H, Ar–H), 6.35 (d, J = 3.8 Hz, 1H, Ar–H), 4.15 (s, 2H, ArNCH2–), 3.91 (m, 2H, –OCHMe2), 3.18 (s, 2H, –NCH2–), 1.27 (d, J = 7.4 Hz, 12H, –CMe2O–), 1.15 (d, J = 6.1 Hz, 12H, –CMe2O–) 13C NMR (CDCl3, 100.63 MHz): δ 159.2, 140.2, 136.7, 115.1, 105.2 (Ar), 79.97 (ArNCH2–), 77.13 (–NCH2–), 75.90 (–OCMe2), 66.01 (–OCMe2), 64.06 (–OCMe2), 28.81 (–OCMe2), 26.06 (–OCMe2), 25.95 (–OCMe2). 13C NMR (CD3OD, 100.63 MHz): δ 157.4, 142.5, 140.4, 113.8, 107.8 (Ar), 72.55 (ArNCH2–), 71.82 (–NCH2–), 65.34 (–OCMe2), 64.74 (–OCMe2), 57.94 (–OCMe2), 27.67 (–OCMe2), 27.65 (–OCMe2), 25.25 (–OCMe2). Anal. calcd for C19H34N2O4Ti: C, 56.72; H, 8.52; N, 6.96. Found: C, 56.84; H, 8.28; N, 7.12.
Synthesis of [LTi(O)]2 (2)
 NCH2–), 2.98 (s, 2H, –NCH2–), 1.26 (s, 6H, –CMe2O–), 1.23 (s, 6H, –CMe2O–). 1H NMR (CD3OD, 400.15 MHz): δ 7.36 (d, J = 7.8 Hz, 1H, Ar–H), 7.23 (m, 1H, Ar–H), 6.64 (d, J = 9.4 Hz, 1H, Ar–H), 6.01 (t, J = 6.7 Hz, 1H, Ar–H), 4.07 (s, 2H, ArNCH2–), 3.07 (s, 2H, –NCH2–), 1.25 (s, 6H, –CMe2O–), 1.21 (s, 6H, –CMe2O–). 13C NMR (CDCl3, 100.63 MHz): δ 156.8, 139.9, 136.3, 113.3, 102.6 (Ar), 71.37 (ArNCH2–), 70.63 (–NCH2–), 64.07 (–OCMe2), 58.90 (–OCMe2), 28.07 (–OCMe2), 27.69 (–OCMe2). 13C NMR (CD3OD, 100.63 MHz): δ 158.2, 142.0, 138.7, 113.5, 105.2 (Ar), 72.63 (ArNCH2–), 72.24 (–NCH2–), 68.86 (–OCMe2), 59.47 (–OCMe2), 27.87 (–OCMe2), 26.49 (–OCMe2). Anal. calcd for C26H41N4O6Ti2: C, 51.93; H, 6.87; N, 9.32. Found: C, 52.19; H, 7.04; N, 9.11.
NCH2–), 2.98 (s, 2H, –NCH2–), 1.26 (s, 6H, –CMe2O–), 1.23 (s, 6H, –CMe2O–). 1H NMR (CD3OD, 400.15 MHz): δ 7.36 (d, J = 7.8 Hz, 1H, Ar–H), 7.23 (m, 1H, Ar–H), 6.64 (d, J = 9.4 Hz, 1H, Ar–H), 6.01 (t, J = 6.7 Hz, 1H, Ar–H), 4.07 (s, 2H, ArNCH2–), 3.07 (s, 2H, –NCH2–), 1.25 (s, 6H, –CMe2O–), 1.21 (s, 6H, –CMe2O–). 13C NMR (CDCl3, 100.63 MHz): δ 156.8, 139.9, 136.3, 113.3, 102.6 (Ar), 71.37 (ArNCH2–), 70.63 (–NCH2–), 64.07 (–OCMe2), 58.90 (–OCMe2), 28.07 (–OCMe2), 27.69 (–OCMe2). 13C NMR (CD3OD, 100.63 MHz): δ 158.2, 142.0, 138.7, 113.5, 105.2 (Ar), 72.63 (ArNCH2–), 72.24 (–NCH2–), 68.86 (–OCMe2), 59.47 (–OCMe2), 27.87 (–OCMe2), 26.49 (–OCMe2). Anal. calcd for C26H41N4O6Ti2: C, 51.93; H, 6.87; N, 9.32. Found: C, 52.19; H, 7.04; N, 9.11.Single crystal X-ray diffraction
Single crystals suitable for X-ray diffraction analysis were obtained from NMR cells containing LH2, 1, and 2 dissolved in CDCl3. The crystallographic measurements were performed at 296(2) K for all compounds LH2, 1, and 2 using a Bruker APEX II diffractometer with Mo Kα (λ = 0.71073 Å) radiation. Specimens of suitable quality and size were selected, coated with Paratone® oil, mounted onto a glass capillary, and centered in the X-ray beam by using a video camera. The structures were solved by the direct method and refined by full matrix least-squares methods using the SHELXTL40 program package with anisotropic thermal parameters for all non-hydrogen atoms. Final refinement based on the reflections (I > 2σ(I)) converged at R1 = 0.0373, wR2 = 0.0872, and GOF = 1.009 for LH2, and at R1 = 0.0322, wR2 = 0.0876, and GOF = 1.021 for 1, and at R1 = 0.0396, wR2 = 0.1022, and GOF = 1.030 for 2. Further details are listed in Table 1. X-ray crystal structures were drawn by the Diamond Program ver. 2.1e.41 Details for crystallographic data and parameters are listed in Table 1.| Sample | LH2 | 1 | 2 |
|---|---|---|---|
| a R1 = Σ||F0| − |Fc||/Σ|F0| and wR2 = {Σ[w(F02 − Fc2)2]/Σ[w(F02)2]}1/2. | |||
| Chemical formula | C13H22N2O2 | C19H34N2O4Ti | C28H42Cl6N4O6Ti2 |
| Formula mass | 238.33 | 402.38 | 839.15 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n | P21/n |
| a (Å) | 12.9760(3) | 10.145(2) | 9.5970(3) |
| b (Å) | 9.0107(2) | 14.8386(3) | 19.6755(6) |
| c (Å) | 12.2487(3) | 14.7511(3) | 9.9008(3) |
| α (°) | 90 | 90 | 90 |
| β (°) | 102.498(2) | 103.9490(10) | 105.588(2) |
| γ (°) | 90 | 90 | 90 |
| Z | 4 | 4 | 2 |
| V (Å3) | 1398.22(6) | 2146.51(8) | 1800.76(10) |
| Temperature (K) | 100(2) | 100(2) | 100(2) |
| Density (g cm−3) | 1.132 | 1.245 | 1.548 |
| μ (mm−1) | 0.077 | 0.423 | 0.934 |
| F(000) | 520 | 864 | 864 |
| θ range (°) | 2.77–25.00 | 1.977–30.689 | 2.070–26.573 |
| Index ranges | −15 ≤ h ≤ 15 | −10 ≤ h ≤ 14 | −12 ≤ h ≤ 12 |
| −10 ≤ k ≤ 10 | −21 ≤ k ≤ 16 | −24 ≤ k ≤ 24 | |
| −14 ≤ l ≤ 13 | −21 ≤ l ≤ 19 | −12 ≤ l ≤ 12 | |
| Reflections collected | 7785 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 010 010 |
31380 |
| # of independent reflections | 2464 | 6620 | 3738 |
| # of parameters | 161 | 272 | 212 |
| GOF(I > 2σ(I)) | 1.009 | 1.021 | 1.030 |
| R1(I > 2σ(I))a | 0.0373 | 0.0322 | 0.0396 |
| wR2(I > 2σ(I))a | 0.0872 | 0.0816 | 0.0955 |
Representative procedures for the cycloaddition reaction of epoxide and CO2
The cycloaddition reaction of CO2 to epoxide was carried out by charging a stirring bar, epoxide (10 mmol), catalyst (20 μmol), and cocatalyst (20 μmol), into a stainless steel pressure reactor (10 mL inner volume). Then, CO2 was charged in the reactor and the pressure was adjusted to desired pressure at appropriate temperature. The reactor was maintained for the desired time. After then, the pressure reactor was cooled to ambient temperature, and the excess CO2 was vented. A small sample of mixture was taken for 1H NMR analysis.Results and discussion
The tridentate ligand HOCMe2CH2NC5H4NCH2CMe2OH (LH2) was synthesized by the reaction of 2-aminopyridine with 2 equivalents of isobutylene oxide in almost quantitative yield. As depicted in Scheme 2, L2H2 was an initial expected product; however, the isolated product from this reaction was LH2, whose the resonance in 6-membered pyridine ring was collapsed. This structure was confirmed by single crystal X-ray analysis (vide infra).
 | ||
| Scheme 1 Synthetic routes for complexes 1 and 2. | ||
 | ||
| Scheme 2 The result of the reaction between 2-aminopyridine and 2 equiv. of isobutylene oxide. | ||
As outlined in Scheme 1, a new monomeric diisopropoxytitanium complex 1 was prepared via simple exchange reaction by adding a solution of Ti(O-i-Pr)4 dropwise to an equimolar solution of LH2 in toluene. Analytically pure samples were obtained as yellow crystalline solids after the removal of the volatiles. Terminal oxo titanium complex 2 can be made by three different synthetic routes as shown in Scheme 1. For Route 1, compound 2 was prepared in situ by treating mixed solution of Ti(O-i-Pr)4 and LH2 with wet air bubbling (Fig. S11 and S12). Wet air bubbling of compound 1 (Fig. S16†) and the direct addition of water into compound 1 (Fig. S17†) were applied for Route 2 and Route 3, respectively. Interestingly, dry air bubbling of compound 1 did not work for the synthesis of compound 2 (Fig. S18†). Thus, oxygen of TiO in compound 2 definitely comes from water. Route 1 is better than Routes 2 and 3 in regards to yield and time efficiency. Unlike compound 1, compound 2 was stable in air. All compounds LH2, 1, and 2 were characterized by 1H and 13C NMR spectroscopies, HR mass spectrometry for LH2 and elemental analysis for 1 and 2 and by single crystal X-ray crystallographic method.
The 1H and 13C NMR spectra were in accord with the suggested structures, and all chemical shifts of the protons and carbons for LH2, 1, and 2 were in the expected range. 1H NMR spectra of 1 and 2 show the absence of signals for alcoholic protons, corroborating successful formation of Ti complexes chelated by L2- ligand. Their molecular structures were determined by single-crystal X-ray diffraction analysis. X-ray quality crystals were obtained by fractional recrystallization at −15 °C in a refrigerator for a few days. Fig. 1–3 display the molecular structures of LH2, 1, and 2, respectively.
 | ||
| Fig. 1 X-ray structure of LH2 with thermal ellipsoids drawn at the 50% probability level. All hydrogens except for alcoholic protons are omitted for clarity. Selected bond lengths (Å): N1–C1 1.366(2); C1–C2 1.352(2); C2–C3 1.411(2); C3–C4 1.351(2); C4–C5 1.442(2); C5–N1 1.402(2); C5–N2 1.296(2); N1–C6 1.474(2); N2–C10 1.455(2). Selected angles (deg): N1–C5–N2 117.10(12); C5–N1–C1 122.00(12); C5–N1–C6 118.57(11); N1–C1–C2 122.17(14); C1–C2–C3 118.33(14); C2–C3–C4 120.57(15); C3–C4–C5 121.79(14); C4–C5–N1 115.03(12); C4–C5–N2 127.87(13); C5–N2–C10 117.56(12). | ||
 | ||
| Fig. 2 X-ray structure of 1 with thermal ellipsoids drawn at the 50% probability level. All hydrogens are omitted for clarity. Selected bond lengths (Å): Ti1–N2 2.2685(9); Ti1–O1 1.8119(8); Ti1–O2 1.8663(8); Ti1–O3 1.8074(9); Ti1–O4 1.8586(8); N1–C1 1.3719(14); C1–C2 1.3520(17); C2–C3 1.4076(18); C3–C4 1.3582(16); C4–C5 1.4358(15); C5–N1 1.3871(14); C5–N2 1.3215(13); C6–N1 1.4685(14); N2–C10 1.4701(14). Selected angles (deg): O1–Ti1–O2 140.83(4); O1–Ti1–O3 106.71(4); O1–Ti1–O4 93.98(4); O2–Ti1–O3 109.73(4); O2–Ti1–O4 89.70(4); O3–Ti1–O4 105.73(4); N2–Ti1–O1 87.29(4); N2–Ti1–O2 76.01(3); N2–Ti1–O3 94.63(4); N2–Ti1–O4 158.25(4); Ti1–O1–C7 152.41(8); Ti1–O2–C11 125.90(7); Ti1–O3–C14 135.37(10); Ti1–O4–C17 130.52(7). | ||
 | ||
| Fig. 3 X-ray structure of 2 with thermal ellipsoids drawn at the 50% probability level. All hydrogens and CDCl3 are omitted for clarity. Selected bond lengths (Å): Ti1–N2 2.153(2); Ti1–O2 2.0549(16); Ti1–O2′ 2.0147(16); Ti1–O1 1.8459(17); Ti1–O3 1.6638(17); N1–C1 1.375(3); C1–C2 1.351(4); C2–C3 1.399(4); C3–C4 1.360(4); C4–C5 1.430(3); C5–N1 1.385(3); C5–N2 1.329(3); N1–C6 1.478(3); N2–C10 1.470(3). Selected angles (deg): O2–Ti1–O2′ 73.50(7); O2–Ti1–O1 139.32(7); O2–Ti1–O3 111.88(8); O2′–Ti1–O1 99.43(7); O2′–Ti1–O3 108.45(8); O1–Ti1–O3 108.35(8); N2–Ti1–O2 77.71(7); N2–Ti1–O2′ 144.99(7); N2–Ti1–O1 89.54(7); N2–Ti1–O3 100.45(8); Ti1–O1–C7 133.55(15). | ||
There are only four π-electrons, not six, in six-membered pyridine ring for tridentate ligand LH2, 1, and 2. As shown in Fig. 4, two different types of carbon–carbon bond lengths certainly exist. A single crystal X-ray diffraction analysis of 1 and 2 confirmed the compounds to be monomeric and dimeric, respectively (Fig. 2 and 3). Complexes 1 and 2 have five-coordinate titanium centers. The titanium atom in 1 is ligated to two O-i-Pr and one tridentate L2−. In addition to anionic oxygens and neutral nitrogen in tridentate L2−, each titanium metal in dimeric 2 has one O ligand and a bridging oxygen from another tridentate L2−, which is the less encumbered tridentate ligand than other tridentate alkoxy/phenoxy pincer ligands bridging two Ti centers.20,42 Two Ti–Obridge bond distances in 2 [2.0549(16) and 2.0147(16) Å] are within the range found in previously reported dimeric Ti complexes having Ti–Obridge bond at 2.0122(16)–2.2332(16) Å.20,42 Interestingly, Ti–O1(terminal) bond length [1.8459(17) Å] is extremely shorter than the two other Ti–Obridge bonds [2.0549(16) and 2.0147(16) Å] in 2. In addition to the short Ti–O1(terminal) bond length, a wide Ti1–O1–C7 bond angle of 133.55(15)° indicates the existence of strong Ti–O π-donation. Thus, O1 atom of L2− chelated to dimeric 2 is somewhat sp2 hybridized and 2 has two weak Ti–Obridge bonds and a strong Ti–O1(terminal) bond. This provides some idea on how the encumbered five coordinate complex 2 could potentially free an open coordination site allowing consequently the epoxide and/or cocatalyst coordination. Complex 2 has an inversion center (i) at the centroid of the Ti2O2 ring plane. There is no direct Ti–Ti interaction for 2.
 | ||
| Fig. 4 Comparison of carbon–carbon bond lengths in 6-membered pyridine ring for LH2, 1, and 2. | ||
The coordination geometry around metal center in the 5-coordinate systems (e.g., trigonal bipyramidal or square pyramidal) could be determined by the trigonality parameter τ (τ = [α − β]/60, where α and β are the largest and next-largest interligand bond angles).43,44 The largest and next-largest interligand bond angles for 1 are ∠O4–Ti1–N2 (158.25(5)°) and ∠O1–Ti1–O2 (140.83(5)°), respectively. Those values for 2 are ∠N2–Ti1–O2′ (145.00(7)°) and ∠O1–Ti1–O2 (139.31(7)°), respectively. Thus, the τ values are 0.29 for 1 and 0.09 for 2. The trigonality parameter τ for the regular trigonal bipyramidal (tbp) complexes is 1.0 and τ for square pyramidal (sqp) complexes is zero. The titanium center in 2 is almost regular square pyramidal geometry; however, 1 could be reasonably defined as having less square pyramidal geometry than 2.
As shown in Fig. 3, compound 2 possesses a square pyramidal geometry where three oxygens and one nitrogen from two L2− ligands occupy the base and oxygen of TiO takes the apical position. The titanium metal is displaced by 0.59 Å toward the oxygen atom of TiO from the base, which is quite planar, the deviations from planarity ranging from −0.396 to 0.347 Å. The TiO bond length in 2 is 1.664(2) Å is within the normal range of structurally characterized terminal oxo-titanium complexes.20–32 Until now, structurally characterized terminal oxo-titanium complexes reported in the literature are all monomeric. In addition, dimeric or polymeric terminal oxo-titanium complexes always use an oxygen atom of TiO as a bridge.33–36 Thus, compound 2 is the first example of dimeric terminal oxo-titanium compound having no TiO→Ti bonds.
The cycloaddition of CO2 to propylene oxide was performed without solvent using the titanium compounds 1 and 2 as catalysts in the presence of cocatalyst. The cycloaddition results are summarized in Table 2. We carried out the reaction at 75 °C under the condition of a fixed [Ti]/[cocatalyst] ratio of 1. Propylene oxide was easily converted into the cyclic carbonate with high selectivity (99%) without any polymerized products. To check the cocatalyst effect on the catalytic activity, we used six different kinds of cocatalysts, such as n-Bu4PBr, n-Bu4NI, n-Bu4NBr, n-Bu4NCl, bis(triphenylphosphoranylidene)ammonium chloride (PPNCl), and 4-dimethylaminopyridine (DMAP). The role of cocatalyst has been demonstrated for several epoxide/CO2 coupling reactions.14–20 It is known that Lewis basic cocatalysts can either (1) be reversibly coordinate to the metal center, increase the electron density on the metal, and labilize the trans ligand to it, acting in turn as nucleophile for the ring opening of epoxide, or (2) the counteranion X− in cocatalyst act as a nucleophile and it proceeds directly to a nucleophilic attack to the coordinated Ti-epoxide. Anionic cocatalysts (n-Bu4NX, n-Bu4PBr, and PPNCl) and non-salt type cocatalyst (DMAP) showed the different catalytic behavior.
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Cocatalyst | Conv. (%) | TONb | Selectivityc (%) |
| a Cycloaddition conditions: [propylene oxide] = 10 mmol, [1] = 0.02 mmol, [2] = 0.01 mmol, [cocatalyst] = 0.02 mmol, [propylene oxide]/[Ti] = 500/1, CO2 = 10 bar, Temperature = 75 °C, time = 24 h.b Turnover number (TON) = (mol of propylene oxide consumed)/(mol of catalyst).c Calculated by 1H NMR spectral integration. | |||||
| 1 | 1 | n-Bu4PBr | 68 | 338 | >99 |
| 2 | n-Bu4NI | 95 | 470 | >99 | |
| 3 | n-Bu4NBr | 78 | 392 | >99 | |
| 4 | n-Bu4NCl | 83 | 416 | >99 | |
| 5 | PPNCl | 66 | 328 | >99 | |
| 6 | DMAP | 21 | 108 | >99 | |
| 7 | 2 | n-Bu4PBr | 69 | 343 | >99 |
| 8 | n-Bu4NI | 51 | 255 | >99 | |
| 9 | n-Bu4NBr | 86 | 432 | >99 | |
| 10 | n-Bu4NCl | 49 | 245 | >99 | |
| 11 | PPNCl | 72 | 362 | >99 | |
| 12 | DMAP | 26 | 132 | >99 | |
| 13 | 1 | None | 20 | 98 | >99 |
| 14 | 2 | 17 | 83 | >99 | |
| 15 | None | n-Bu4PBr | 11 | 54 | >99 |
| 16 | n-Bu4NI | 13 | 64 | >99 | |
| 17 | n-Bu4NBr | 12 | 59 | >99 | |
| 18 | n-Bu4NCl | 10 | 49 | >99 | |
| 19 | PPNCl | 13 | 64 | >99 | |
| 20 | DMAP | 5 | 24 | >99 | |

As expected, cocatalyst n-Bu4NI for catalyst 1 is superior compared with the five others (entries 1–6) because the nucleophilicity for halide ions decreases on going up from I− to Cl−. However, no similar trend for catalyst 2 was observed and the highest catalytic activity for 2 was obtained in the presence of n-Bu4NBr (entry 9). Interestingly, amine-based cocatalyst such as n-Bu4NBr showed the better activity than phosphine-based cocatalyst, n-Bu4PBr in both 1 and 2 (entries 1 and 3 for 1 and entries 7 and 9 for 2). The reason may be quaternary ammonium compounds could stabilize the carbonate intermediate. Compounds 1 and 2 showed marginal difference in catalytic activity (entries 1–12). Both 1 and 2 with non-salt-type cocatalyst DMAP showed very low activities (entries 6 and 12). Interestingly, compounds 1 and 2 without any cocatalyst showed the conversion of 20% and 17%, respectively, under the same cycloaddition condition (entries 13 and 14). In addition, six cocatalysts without 1 and 2 showed low conversion in the range of 5–13% (entries 15–20). Thus the coupling of catalyst and cocatalyst showed the synergistic activity for this cycloaddition reaction.
Until now, four papers on Ti-based systems for the cycloaddition of CO2 and epoxides to make cyclic carbonates were reported in the literature.14–17 Harsh condition such as high catalyst loading of 1 mol%,14,15,17 high temperature such as 100 °C (ref. 17) and 150 °C (ref. 14 and 15) and high CO2 pressure such as 15 bar (ref. 17) and 22 bar (ref. 16) for the reported systems is needed. However, our system was applied under the condition of 0.2 mol% catalyst loading, 75 °C, and 10 bar CO2 pressure. Even though all systems conducted in different reaction condition, our systems are one of the mildest Ti-based systems. Interestingly, compounds 1 and 2 (0.2 mol%) showed complete conversion at 125 °C within 1 h. In addition, catalyst/cocatalyst ratio of 2 and CO2 pressure change did not show any significant differences. Interestingly, in case of the synthesis of polymers via coupling reaction of propylene oxide and styrene oxide with CO2 catalyzed by homogeneous Ti-based complexes under mild or similar conditions (1–35 bar of CO2 pressure and 25–80 °C).18–20
We next moved to investigate the scope of substrates (Table 3) using 1 as a catalyst and n-Bu4NI as a cocatalyst. Substrates include various kinds of epoxides such as propylene oxide (Table 2, entry 1), 1,2-epoxybutane (entry 2), 1,2-epoxyhexane (entry 3), epichlorohydrin (entry 4), 1,2-epoxy-3-methoxypropane (entry 5), tert-butyl glycidyl ether (entry 6), and 1,2-epoxy-2-methylpropane (entry 7). Epoxides with different alkyl substituents (R = Me, Et, and n-Bu) showed high dependence on chain length (entries 1–3). While propylene oxide gave an excellent conversion (entry 1), a quick loss of reactivity was observed from ethyl to butyl side chains (entries 2 and 3). Interestingly, when heteroatom was introduced at 3-position, epichlorohydrin (entry 4) showed the better activity than epoxides with –CH2OMe (entry 5) and –CH2OBut (entry 6) substituents. However, substrate with multi-substituents such as 1,2-epoxy-2-methylpropane (entry 7) exhibited low conversions. The observed dependency of the nature of the epoxide and the observed TONs may be related to the polarity of the reaction mixture and thus the solubility of the catalyst species could have been influenced.
|
||||
|---|---|---|---|---|
| Entry | Product | Time (h) | TONb | Selectivityc (%) |
| a Cycloaddition conditions: [epoxide] = 10 mmol, [1] = 0.02 mmol, [n-Bu4NI] = 0.02 mmol, [epoxide]/[1] = 500/1, CO2 = 10 bar, temperature = 75 °C.b Turnover number (TON) = (mol of epoxide consumed)/(mol of catalyst).c Calculated by 1H NMR spectral integration. | ||||
| 1 |  |
24 | 470 | >99 |
| 2 | 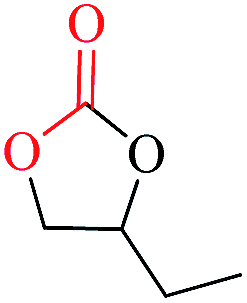 |
48 | 344 | >99 |
| 3 |  |
48 | 250 | >99 |
| 4 |  |
14 | 403 | >99 |
| 5 |  |
24 | 330 | >99 |
| 6 |  |
46 | 406 | >99 |
| 7 |  |
54 | 74 | >99 |

Conclusions
The ligand LH2 from the reaction between 2-aminopyridine and 2 equiv. of isobutylene oxide was synthesized and used to make monomeric LTi(O-i-Pr)2 (1) and dimeric [LTi(O)]2 (2). Compound 2 has terminal oxo group and was bridged by the oxygen atom of L2− ligand. Compound 2 is the first example of terminal oxo-titanium compound having no TiO→Ti bonds. New titanium complexes 1 and 2 could act as catalysts for the synthesis of cyclic carbonate via the cycloaddition of CO2 to epoxides under somewhat mild condition of 0.2 mol% catalyst loading, 75 °C, and CO2 pressure of 10 bar compared with other reported Ti-based catalysts.
Acknowledgements
Y. K. thanks the National Research Foundation of Korea (NRF), the Korean Ministry of Education (MOE) through the Creative Human Resource Training Project for Regional Innovation (grant number 2014H1C1A1066874) and Basic Science Research Program (grant number 2015R1D1A1A01061043) for financial support. J. L. acknowledges financial supports by the BRL Program (2015R1A4A1041036) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning.Notes and references
- E. Lichtfouse, J. Schwarzbauer and D. Robert, CO2 Sequestration: Biofuels and Depollution, Springer, Switzerland, 2015 Search PubMed.
- J. Bayardon, J. Holz, B. Schäffner, V. Andrushko, S. Verekin, A. Preetz and A. Börner, Angew. Chem., Int. Ed., 2007, 46, 5971–5974 CrossRef CAS PubMed.
- M. Arakawa, S. Tobishima, T. Hirai and J. Yamaki, J. Electrochem. Soc., 1986, 133, 1527–1528 CrossRef CAS.
- T. Takata, Y. Furusho, K. Murakawa, T. Endo, H. Matsuoka, T. Hirasa, J. Matsuo and M. Sisido, J. Am. Chem. Soc., 1998, 120, 4530–4531 CrossRef CAS.
- K. Biggadike, R. M. Angell, C. M. Burgess, R. M. Farrell, A. P. Hancock, A. J. Harker, W. R. Irving, C. Ioannou, P. A. Procopiou, R. E. Shaw, Y. E. Solanke, O. M. P. Singh, M. A. Snowden, R. J. Stubbs, S. Walton and H. E. Weston, J. Med. Chem., 2000, 43, 19–21 CrossRef CAS PubMed.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- S. Narang, R. Mehta and S. N. Upadhyay, Curr. Org. Chem., 2015, 19, 2344–2357 CrossRef CAS.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- M. North, Sustainable Catalysis: With Non-endangered Metals, Part 1, Royal Society of Chemistry, 1st edn, 2015, ch. 5–7 Search PubMed.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007 RSC.
- E. Le Roux, Coord. Chem. Rev., 2016, 306, 65 CrossRef CAS.
- D. Bai, H. Jing, Q. Liu, Q. Zhu and X. Zhao, Catal. Commun., 2009, 11, 155–157 CrossRef CAS.
- D. Bai, G. Nian, G. Wang and Z. Wang, Appl. Organomet. Chem., 2013, 27, 184–187 CrossRef CAS.
- M. J. Go, K. M. Lee, C. H. Oh, Y. Y. Kang, S. H. Kim, H. R. Park, Y. Kim and J. Lee, Organometallics, 2013, 32, 4452–4455 CrossRef CAS.
- F. Al-Qaisi, E. Streng, A. Tsarev, M. Nieger and T. Repo, Eur. J. Inorg. Chem., 2015, 5363–5367 CrossRef CAS.
- M. Mandal and D. Chakraborty, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 809–824 CrossRef CAS.
- K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed.
- J. Hessevik, R. Lalrempuia, H. Nsiri, K. W. Törnroos, V. R. Jensen and E. Le Roux, Dalton Trans., 2016, 45, 14734–14744 RSC.
- P. N. Dwyer, L. Puppe, J. W. Buchler and W. R. Scheidt, Inorg. Chem., 1975, 14, 1782–1785 CrossRef CAS.
- R. Guilard, J. M. Latour, C. Lecomte, J. C. Marchon, J. Protas and D. Ripoll, Inorg. Chem., 1978, 17, 1228–1237 CrossRef CAS.
- R. Crescenzi, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1996, 15, 5456–5458 CrossRef CAS.
- R. Crescenzi, E. Solari, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1999, 18, 606–618 CrossRef CAS.
- S.-H. Hsu, J.-C. Chang, C.-L. Lai, C.-H. Hu, H. M. Lee, G.-H. Lee, S.-M. Peng and J.-H. Huang, Inorg. Chem., 2004, 43, 6786–6792 CrossRef CAS PubMed.
- T. E. Hanna, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2007, 46, 2359–2361 CrossRef CAS PubMed.
- M. R. Smith, P. T. Matsunaga and R. A. Andersen, J. Am. Chem. Soc., 1993, 115, 7049–7050 CrossRef CAS.
- T. T. Nguyen, G. D. Kortman and K. L. Hull, Organometallics, 2016, 35, 1713–1725 CrossRef CAS.
- J. E. Hill, P. E. Fanwick and I. P. Rothwell, Inorg. Chem., 1989, 28, 3602–3604 CrossRef CAS.
- A. Bodner, P. Jeske, T. Weyhermüller, K. Wieghardt, E. Dubler, H. Schmalle and B. Nuber, Inorg. Chem., 1992, 31, 3737–3748 CrossRef CAS.
- P. Jeske, G. Haselhorst, T. Weyhermüller, K. Wieghardt and B. Nuber, Inorg. Chem., 1994, 33, 2462–2471 CrossRef CAS.
- J. R. Hagadorn and J. Arnold, Organometallics, 1998, 17, 1355–1368 CrossRef CAS.
- D. P. Solowey, T. Kurogi, B. C. Manor, P. J. Carroll and D. J. Mindiola, Dalton Trans., 2016 10.1039/c6dt01534d.
- F. Franceschi, E. Gallo, E. Solari, C. Floriani, A. Chiesi-Villa, C. Rizzoli, N. Re and A. Sgamellotti, Chem.–Eur. J., 1996, 2, 1466–1476 CrossRef CAS.
- A. J. Nielson and J. M. Waters, Eur. J. Inorg. Chem., 2015, 2028–2032 CrossRef CAS.
- M. M. Olmstead, P. P. Power and M. Viggiano, J. Am. Chem. Soc., 1983, 105, 2927–2928 CrossRef CAS.
- G. R. Willey, J. Palin and M. G. B. Drew, J. Chem. Soc., Dalton Trans., 1994, 1799–1804 RSC.
- D. F. Shriver and M. A. Drezdzon, The Manipulation of Air-Sensitive Compounds, Wiley, New York, 2nd edn, 1986 Search PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Elsevier, New York, 6th edn, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- DIAMOND version 2.1c, Crystal Impact GbRBonn, Germany, 1999 Search PubMed.
- S. R. Golisz, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 5026–5032 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. H. Kim, S. Y. Han, J. H. Kim, Y. Y. Kang, J. Lee and Y. Kim, Eur. J. Inorg. Chem., 2015, 2323–2329 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: crystallographic data in CIF and 1H and 13C NMR for LH2, 1, 2, and cyclic carbonates synthesized. CCDC 1497957–1497959. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra23255h |
| ‡ The first and second authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2016 |