
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High performance stability of titania decorated carbon for desalination with capacitive deionization in oxygenated water†
Pattarachai Srimukab,
Lucie Riesab,
Marco Zeigerab,
Simon Fleischmannb,
Nicolas Jäckelab,
Aura Tolosaab,
Benjamin Krünerab,
Mesut Aslana and
Volker Presser*ab
aINM – Leibniz Institute for New Materials, Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, Saarbrücken, Germany
First published on 1st November 2016
Abstract
Performance stability in capacitive deionization (CDI) is particularly challenging in systems with a high amount of dissolved oxygen due to rapid oxidation of the carbon anode and peroxide formation. For example, carbon electrodes show a fast performance decay, leading to just 15% of the initial performance after 50 CDI cycles in oxygenated saline solution (5 mM NaCl). We present a novel strategy to overcome this severe limitation by employing nanocarbon particles hybridized with sol–gel-derived titania. In our proof-of-concept study, we demonstrate very stable performance in low molar saline electrolyte (5 mM NaCl) with saturated oxygen for the carbon/metal oxide hybrid (90% of the initial salt adsorption capacity after 100 cycles). The electrochemical analysis using a rotating disk electrode (RDE) confirms the oxygen reduction reaction (ORR) catalytic effect of FW200/TiO2, preventing local peroxide formation by locally modifying the oxygen reduction reaction.
Introduction
Capacitive deionization (CDI) is an emerging water treatment technology which is highly attractive for energy efficient removal of ions from aqueous media with low molar concentrations (especially below 100 mM).1–3 Common CDI operation accomplishes ion removal from a feed stream via capacitive ion electrosorption at the electrically charged fluid/solid interface between the electrolyte and the electrode. Most commonly, CDI employs nanoporous carbon, such as activated carbon, but also other carbons have been explored, for instance, graphene, carbon nanotubes, or carbide-derived carbon.4 During electrical charging of the carbon electrodes, ions are removed from the feed stream via electrosorption.5 A key value for CDI benchmarking is the desalination capacity (SAC = salt adsorption capacity), normalized to the electrode mass, and values of 15–21 mg g−1 have been reported.6,7CDI performance is commonly plagued by progressing degradation stemming from the electrochemical deterioration of the carbon electrodes.8–10 A challenge is the aggravating difference between the positively and negatively charged electrode: the former is continuously oxidized, leading to a shift in the electrode potential towards the oxygen reduction reaction (ORR) limit and enhanced degradation.9,11 One approach to mitigate the issue is the use of dissimilar electrodes.12,13 For example, modifying the carbon surfaces with functional groups allows to introduce an additional chemical charge and, effectively, shifts the point of zero charge of the respective electrodes.14,15 By this way, CDI can even be completely inverted to yield ion release upon charging and desalination during discharging. Yet, the main focus of research on tuning carbon surface charge and functionality has remained on enhancing the salt removal capacity or the efficiency, but the longevity and performance stability remain widely unexplored. This is even more aggravated for practical, non-ideal systems, that is, aqueous saline media with dissolved oxygen, where H2O2 evolution occurs on the negatively polarized electrode.16,17
Besides other metal oxides (like manganese oxide18 or zinc oxide19), titania/carbon hybrid materials have been investigated to enhance the desalination capacity20,21 and/or efficiency.22,23 These studies commonly cover the range of few desalination/regeneration (charge/discharge) cycles, but even for CDI with just carbon electrodes, only few works show dozens or more cycles.8 So far, research has fallen short of investigating the potential for enhancing the CDI performance stability by use of metal oxide coatings.
In this study, we explore the hybridization of few-nanometer-sized carbon black particles (FW200) with titania via sol–gel synthesis and report on the remarkable performance stability in low molar saline media with a high amount of dissolved oxygen. We chose carbon black instead of microporous activated carbon because we wanted to avoid ion transport limitations within the network of nanopores in micrometer sized particles and to have access for titania decoration to an exclusive outer surface area. We also study the ORR catalyst ability of FW200 and FW200/TiO2 to understand the origin of longevity performance in harsh (i.e., oxygenated) solution.
Experimental
Material synthesis
For our study, we used carbon black type FW200, which was purchased from Orion (formerly: Degussa) and used without further chemical treatment.For FW200/TiO2 synthesis, 18 g of vacuum dried carbon powder FW200 was dispersed in 200 mL ethanol and set on magnetic stirrer for 5 h. Then, 16.1 g Ti(IV) isopropoxide (Aldrich) was dissolved in 500 mL ethanol and set on magnetic stirrer which should give a carbon hybrid with 20 mass% titania. During filling, the bottles/containers were purged with argon to avoid any moisture pick up from the environment. The Ti-isopropoxide solution and carbon slurry were poured together and stirred for further 48 h without any additional water for hydrolysis. After the surface functional groups of FW200 have reacted with Ti-isopropoxide, the slurry was centrifuged at 4000 rpm for 1 h so that the excess of unreacted Ti-isopropoxide was removed. The sediment was dispersed in 500 mL ethanol and centrifuged and the supernatant was removed again. This procedure was repeated two times to remove the unreacted Ti-isopropoxide. The cleaned sediment was dried at 95 °C for 24 h and used for the electrode preparation.
Structural and chemical characterization
Scanning electron microscope (SEM) images were recorded with a JEOL JSM 7500F field emission scanning electron microscope operating at 3 kV. Energy dispersive X-ray spectroscopy (EDX) was carried out in the system with an X-Max silicon detector from Oxford Instruments using AZtec software for quantitative analysis. Transmission electron micrographs were taken with a JEOL 2100F transmission electron microscope at 200 kV. Powder samples were prepared by dispersing the powder in ethanol and drop casting them on a copper grid with lacey carbon film (Gatan).Raman spectra were recorded with Renishaw inVia Raman microscope using a Nd-YAG laser with an excitation wavelength of 523 nm. The spectral resolution was 1.2 cm−1 and the diameter of laser spot on the sample was 2 μm with a power of 0.2 mW. The spectra were recorded for 20 s and accumulation of 30-times to get high signal-noise and signal-background ratio.
X-ray diffraction (XRD) was conducted per use of a D8 Advance diffractometer (Bruker AXS) with a copper X-ray source (CuKα, 40 kV, 49 mA) in point focus (0.5 mm) and a Goebel mirror. A VANTEC-500 (Bruker AXS) 2D detector was employed (25° per step: measurement time 16.7 min per step). The sample was dispersed in ethanol and drop casted on a sapphire wafer and the sample holder was oscillating horizontally to enhance statistics with an amplitude of 5 mm with speed of 0.5 mm s−1 along the x- and 0.2 mm s−1 along the y-axis.
Nitrogen gas sorption measurements at −196 °C were carried out with an Autosorb system (Autosorb 6B, Quantachrome). The powder samples were outgassed at 250 °C for 10 h under vacuum conditions at 102 Pa to remove adsorbed water; the outgassing temperature for film electrodes was 120 °C. Nitrogen gas sorption was performed in liquid nitrogen in the relative pressure range from 0.008 to 1.0. The Brunauer–Emmett–Teller specific surface area (BET-SSA)24 was calculated with the ASiQwin-software in the linear relative pressure range of 0.06–0.1. The density functional theory specific surface area (DFT-SSA) and pore size distribution were calculated via quenched-solid density functional theory25 assuming slit-shaped pores.
Thermogravimetric analysis was carried out on dried hybrid samples at 10 °C min−1 to 900 °C in flowing synthetic air (20 cm3 min−1) to determine the ash content, which corresponds to the total TiO2 content in the samples (TG 209F1, Netzsch).
Electrode preparation
All film electrodes were casted on graphite current collectors using a mixed PVP/PVB polymer binder. We had shown the stability of this binder system in aqueous NaCl solutions in a previous publication26 and the group of Likun Pan established CDI compatibility when using this binder in a recent study.27 The carbon hybrid slurry was prepared by dispersing 5 g of as-prepared material and 128 mg of polyvinylpyrrolidone (PVP) in 20 g of ethanol. After tip sonication for 10 min, 319 mg of polyvinyl butyral (PVB; 25 mass% in ethanol) was added and stirred for 30 min. The solid content of pure carbon slurry was set to 10 mass% to ensure castability. Afterwards, the slurry was casted on SGL graphite current collectors (250 μm thickness) and dried at 80 °C for 24 h. The thickness of tape casted electrodes was 100 μm for FW200 and 150 μm and for FW200/TiO2.Electrochemical measurements
We prepared electrodes by using the same method as introduced for CDI electrodes. The slurry was drop casted on the graphite current collector. The as-prepared electrodes with a diameter of 10 mm were placed into our custom-build cell having spring loaded titanium pistons. A glass fiber mat (GF/A, Whatman) was used as a separator. 1 M NaCl was injected by vacuum backfilling. Electrochemical measurement was conducted with a VSP300 potentiostat/galvanostat (Bio-Logic). For the symmetric full-cell, cyclic voltammetry and galvanostatic cycling techniques were employed. The specific capacitance of the cell was calculated by using eqn (1):
 | (1) |
To monitor the potential development of counter and working electrode in a two electrode setup, we introduced a Ag/AgCl spectator reference electrode to the cell. During constant specific current of 0.1 A g−1 (charge/discharge), the cathode and anode potential were monitored by reference electrode and the different of potentials between cathode and anode were determined.
Oxygen reduction reaction (ORR) evaluation
A rotating disk electrode (RDE) was used to investigate ORR for FW200 and FW200/TiO2. The RDE working electrode is a glassy carbon (GC) electrode (Bioanalytical Systems) with a diameter of 5 mm. Prior to use, the RDE-GC was polished on pat with alumina powder slurry several times and dried at 60 °C for 6 h. The carbon hybrid slurry was prepared by the following steps. Active carbon hybrid material (FW200 or FW200/TiO2) was mixed with 2.5 mg of PVP in 5 mL absolute ethanol. After that, 23 mg of PVB solution (25 mass% ethanol) was added to the previous mixture and stirred for 15 min. The paste was drop casted on RDE-GC (3 μL of slurry) and dried at 60 °C for 2 h. The as prepared RDE was connected to the RDE apparatus (RRDE-3A, Bioanalytical Systems) having a Pt wire as counter electrode and Ag/AgCl (saturated KCl) as reference electrode. Prior to ORR analysis, 1 M NaCl was bubbled with O2 gas for 20 min. Linear sweep voltammograms were applied at a sweep rate of 10 mV s−1 to investigate the ORR on both the FW200 and FW200/TiO2 electrode. The working electrode potential was scanned from 0.2 to −0.6 V vs. Ag/AgCl. The rotating speed was varied from 0 rpm to 3200 rpm. To subtract the background capacitive current, RDE was tested in an O2-free solution at 0 rpm.The Koutecky–Levich equation (eqn (2) and (3)) was used to estimate the number of electron transfers:28
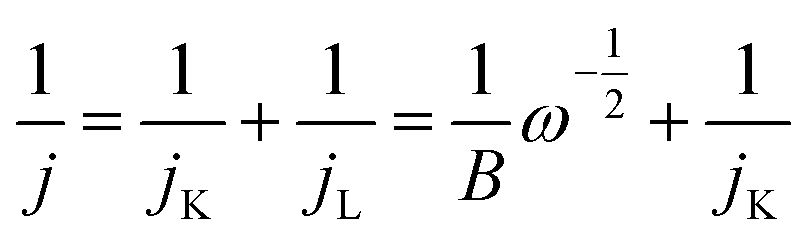 | (2) |
 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), CO is the concentration of oxygen in 1 NaCl at 25 °C 1 atm (2.59 × 10−7 mol cm−3),31 and n is the number of electron transfers involved in ORR. The parameter jK is assumed to be constant after reaching equilibrium potential at one specific mass transfer condition (jL). As shown in eqn (2), the measured current exhibits linear relation with ω−1/2; therefore, the inverse portion of B indicates the slope of K–L plot which allows the calculation of n per use of eqn (3).
485 C mol−1), CO is the concentration of oxygen in 1 NaCl at 25 °C 1 atm (2.59 × 10−7 mol cm−3),31 and n is the number of electron transfers involved in ORR. The parameter jK is assumed to be constant after reaching equilibrium potential at one specific mass transfer condition (jL). As shown in eqn (2), the measured current exhibits linear relation with ω−1/2; therefore, the inverse portion of B indicates the slope of K–L plot which allows the calculation of n per use of eqn (3).
CDI measurements
The CDI setup described in ref. 32 with flow-by electrodes (per definition in ref. 1) was used to characterize the desalination performance. The CDI stack was built from the as prepared electrodes and a porous spacer (glass fiber pre-filter, Millipore, 380 μm thickness). The measurements were carried out with three pairs of electrodes. Ion adsorption and desorption steps were carried out using constant potential mode at 1.2 V. The electrode regeneration was accomplished at 0 V. For all electrochemical operations, we used a VSP300 potentiostat/galvanostat (Bio-Logic) and the duration of each half-cycle was 30 min. All experiments were carried out with a flow rate of 22 mL min−1 of 5 mM NaCl solution and a 10 L electrolyte tank which was flushed continuously with O2 gas to ensure oxygen saturation. The salt adsorption capacity (SAC) and the measured charge were defined per mass of active material in both electrodes. For quantification of the electrical charge, the leakage current measured at the end of each half-cycle was subtracted.Results and discussion
Structural and chemical properties
Carbon black type FW200 exhibits micrometer-sized aggregates (Fig. 1A and B), which consist of nanometer-sized primary particles of around 5–15 nm containing highly disordered carbon, as shown by transmission electron microscopy (Fig. 1C). The small size of the carbon black particles was intentionally chosen for providing facile access to the pore volume available for CDI operation and to minimize transport limitations due to impeded ion diffusion. The sol–gel synthesis of titania applied in our experiments yielded a homogenous hybridization of the carbon material (Fig. 1A), as can be seen from the elemental mapping of titanium (Fig. 1B). Quantitative analysis for the EDX spectra (Fig. 1D) yielded for FW200 91 ± 1 mass% carbon and 9 ± 1 mass% oxygen, and for FW200/TiO2 a composition of 85 ± 1 mass% C, 13 ± 1 mass% O, and 2 ± 1 mass% Ti. High resolution transmission electron microscopy revealed small clusters (ca. 1–2 nm) of titania (Fig. 1C, inset). The hybrid powder showed a mass loading of ca. 8 mass% titania (=4.8 mass% titanium) as confirmed by thermogravimetric analysis (Fig. 1E). That value is larger than the amount of Ti determined by EDX (2 mass%), because we have to consider the small spot size of the electron beam (effectively probing a small μm3 volume) and the thermogravimetric analysis is more representative for the total titanium mass in the sample. We see in the thermogram also an onset of the carbon oxidation at much lower temperatures for FW200/TiO2 compared to just FW200. This is explained by the catalytic effect of the metal oxide on the carbon oxidation reaction.33 | ||
| Fig. 1 (A) Scanning electron micrograph and (B) overlaid titanium chemical map (EDX) of hybrid FW200/TiO2. (C) Transmission electron micrographs of FW200/TiO2 showing small titania domains. (D) EDX spectra of FW200 and FW200/TiO2 along with quantitative data for C, O, and Ti. (E) Thermogravimetric analysis in synthetic air of FW200 and FW200/TiO2. (F) Raman spectra and (G) X-ray diffractograms of FW200 and FW200/TiO2. | ||
As seen from the Raman spectra (Fig. 1F), FW200 and FW200/TiO2 display the typical pattern of carbon which consists of the D-band and the G-band at the wavelengths of about 1354 cm−1 and 1603 cm−1 and a distinct second order spectrum between 2200 and 3200 cm−1.34 The spectra of FW200 and FW200/TiO2 are virtually indistinguishable (Fig. 1F). We also did not observe any characteristic peaks of titania in FW200/TiO2 since the domain size of titania (ca. 2 nm; Fig. 1B) is too small to yield a detectable Raman signal. X-ray diffraction also shows only the presence of carbon per the peaks at 25.5° 2θ and 42° 2θ, corresponding with the (002) and (110) reflections of graphitic carbon (Fig. 1G). This, too, is in alignment with the presence of few-nanometer-sized titania domains, scattered throughout the network of FW200 particles.
Adding metal oxide to carbons may result in pore blocking of the electrode.35 By using small amounts of titania and small particles, we were able to limit porosity reduction (Fig. 2). In particular, we see a reduction of the DFT surface area from 549 m2 g−1 of FW200 to a value of 404 m2 g−1 for FW200/TiO2 (i.e., −27%). This decrease can already be assessed from the nitrogen sorption isotherms (Fig. 2). Both isotherms are consistent with a material with interparticle nanopores like carbon onions.36 The measured surface areas align with the presence of pores just in-between the FW200 grains, with an ideal surface area of up to 545 m2 g−1 for non-porous carbon spheres of 5–15 nm diameter. Concluding from quenched solid density functional theory, the corresponding decrease in surface area is mostly accomplished by a decrease in micropores (i.e., pores below 2 nm) as a result from certain pore blocking by titania nano-domains (Fig. 2B). The higher density of the metal oxide also contributes towards the reduction of surface area.
 | ||
| Fig. 2 (A) Nitrogen gas sorption isotherm of FW200 and FW200/TiO2 showing pore blocking for the latter. (B) Differential pore size distribution calculated via quenched solid density functional theory assuming slit-shaped pores for FW200 and FW200/TiO2 showing the mixed micro-mesoporous nature of both materials. | ||
Electrochemical characterization in high ionic strength
The full-cell electrochemical measurements were carried out in oxygen free 1 M NaCl solution to investigate the charge adsorption capability of FW200 and FW200/TiO2. As seen in Fig. 3A, the cyclic voltammograms of FW200 and its composite at 5 mV s−1 exhibit rectangular shape, indicative of charge storage predominately accomplished by double-layer formation (i.e., ion electrosorption).37 The same is indicated by the pronouncedly triangular shape of galvanostatic charge–discharge plots (Fig. 3B). The specific capacitance of FW200 and FW200/TiO2 at low specific current of 0.1 A g−1 is 112 F g−1 and 101 F g−1, respectively, while the specific capacitance at high specific current of 10 A g−1 is 43 F g−1 for FW200 and 47 F g−1 for FW200/TiO2, respectively (Fig. 3C). The lower specific surface area of FW200/TiO2 accounts for the lower specific capacitance, although the difference is much less as might be indicated from the 27% lower accessible surface area compared to FW200. | ||
| Fig. 3 (A) Cyclic voltammograms at 5 mV s−1, (B) galvanostatic charge–discharge curve at 0.1 A g−1, (C) rate capability, and (D) cathode, anode and zero charge potential monitoring along with different cell voltages. | ||
The potential development of cathode and anode in the full-cell was benchmarked by introducing an Ag/AgCl spectator electrode (Fig. 3D). The zero charge potentials (E0) of FW200 and FW200/TiO2 are shifted to positive values vs. Ag/AgCl at all studied cell voltages (i.e., between 0.6 V and 1.4 V). This implies the influence of negatively charged surface groups which get neutralized by cations, leading to an asymmetric potential distribution between cathode (ΔEcathode = 0.64 V vs. Ag/AgCl at cell voltage of 1.2 V) and anode (ΔEanode = −0.56 V vs. Ag/AgCl at cell voltage of 1.2 V). Nanodecoration with titania further shifts the potentials to positive values of Δ80 mV at 0.6 V cell voltages; yet, the differences gradually reduce and virtually vanish at 1.4 V.
Capacitive deionization performance
The desalination performance of FW200 and FW200/TiO2 was measured at 1.2 V cell voltage, using a symmetrical two-electrode setup and aqueous 5 mM NaCl saline solution. We bubbled the electrolyte with O2 to achieve oxygen saturation. This is in stark contrast to the majority of work in the CDI literature, where, with some exceptions (e.g., ref. 38), mostly de-aerated saline media are investigated.1,2 FW200 carbon electrodes required 35 CDI cycles (i.e., charging and discharging cycles) to achieve equilibrium performance, that is, to obtain the same salt adsorption and salt desorption capacity. Only after such equilibrium is reached, meaningful values for the salt adsorption capacity (SAC) can be obtained and compared to literature.1 During the 35 conditioning cycles, there were strong fluctuations of the online monitored conductivity of the out-flowing saline solution. We even observed, limited to the early cycles during conditions, evidence of CDI inversion, where ions are desorbed during charging (Fig. 4A).9,10,38 These high fluctuations are a result of the nanoscopic size of the carbon black primary particles and the obvious reactivity with surface functional groups, which are evidenced by an oxygen content of 9 mass% in FW200, as measured by EDX. After the initial conditioning, conventional CDI adsorption/desorption cycles occurred for FW200, as depicted in Fig. 4B (highlighting the 10th CDI cycle). Prolonged operation of FW200, however, lead to a gradual decrease of the SAC values. Starting from 10 ± 2 mg g−1 for the first CDI cycle (averaged over two experiments), the performance decrease follows almost an exponential law and fades by 80% to ca. 2 mg g−1 after 20 CDI cycles and by 90% to ∼1 mg g−1 after 60 CDI cycles.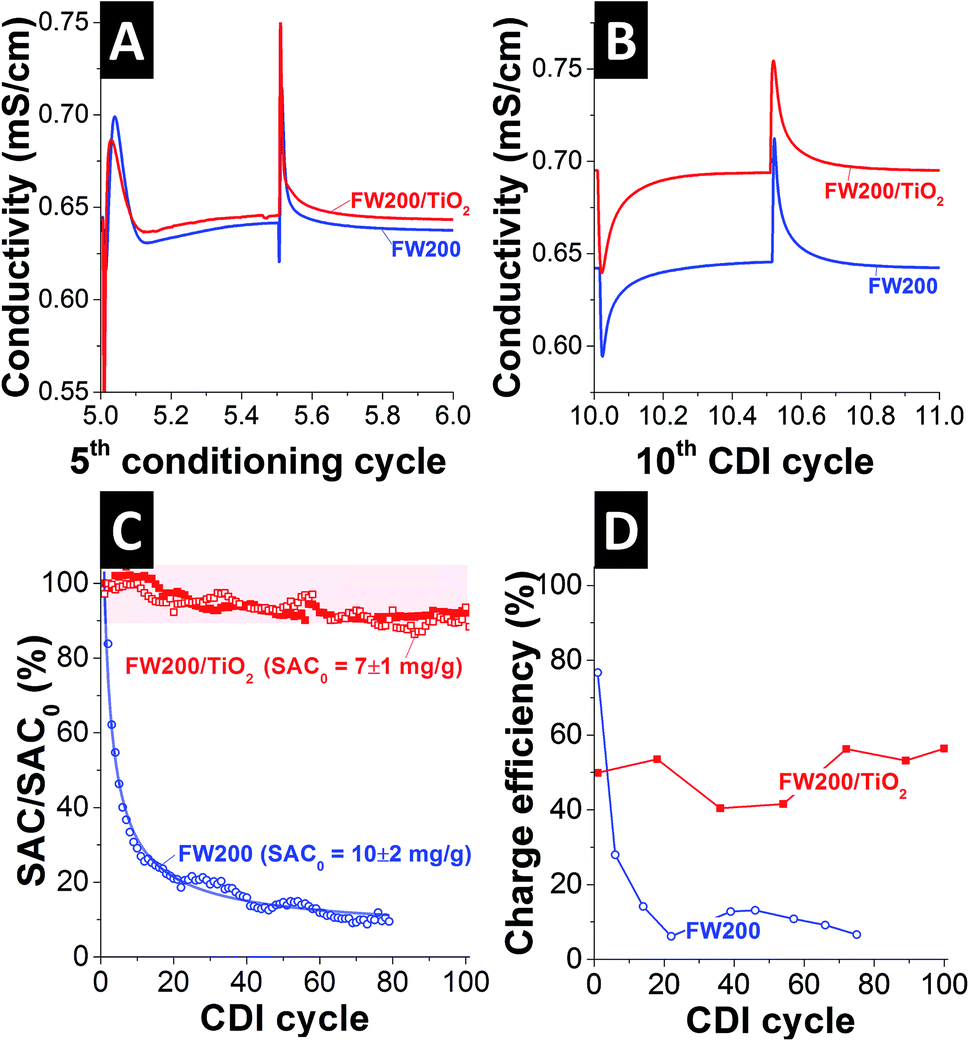 | ||
| Fig. 4 (A) Typical conductivity curve for a conditioning cycle (shown data for the 5th conditioning cycle) with inverted CDI operation during the desorption cycle (1st half cycle). (B) Typical conductivity curve for conditioned CDI operation (shown data for the 10th CDI cycle) with regular ad- and desorption half-cycles. (C) Salt adsorption capacity (SAC) performance normalized to the first-cycle CDI cycle. The solid lines are fits for an exponential decay (blue; for FW200). For FW200/TiO2, two data sets of individual CDI cells are presented. (D) Charge efficiency values corresponding with the data shown in panel (C). | ||
In case of FW200/TiO2, we also observed an initial conditioning phase of 35 cycles (Fig. 4A), following the same pattern as FW200 (Fig. 4B). Obviously, the run-in behavior is dominated by the majority phase (i.e., FW200 = 92 mass%) and its surface chemistry. Yet, the initial SAC of the first CDI cycle (i.e., after conditioning) of 7 ± 1 mg g−1 (averaged over three experiments) reduces by only ca. 10% after 100 CDI cycles (Fig. 4C). To the best of our knowledge, this is by far the highest performance stability for any CDI system reported so far for oxygen-saturated saline electrolyte without the use of membranes. We also highlight that we used a cell voltage of 1.2 V, instead of a lower voltage, as surveyed for example in ref. 9. The latter reference illustrates the benefit in performance stability when reducing the cell voltage, for example, from 0.9 V to 0.7 V. The observed performance stability enhancement is clearly linked with the presence of titania. When using standard carbon materials, dissolved oxygen is reduced and consumed to form H2O2 as the major reagent during CDI.39 Once H2O2 is formed, the oxidation of carbon is boosted and this leads to severe degradation of the carbon electrode material.16,17 The mechanism for oxygen reduction reaction over carbon material in alkaline media has been reported elsewhere.40
Charge efficiency, the ratio between invested charge and removed ions, is a useful tool to further characterize CDI performance and stability.41 As can be seen from Fig. 4D, the initial charge efficiency for FW200 is high with ca. 80%, but drops to ca. 10% after 20 cycles. This aligns with the fast decay in SAC, as seen in Fig. 4C, and low values for the charge efficiency are common for carbons with a high heteroatom content in the form of surface functionalities.42 The titania decorated hybrid electrode displays a lower, but much more stable charge efficiency with an average value of 50 ± 6% over 100 cycles (Fig. 4D). The lower value may result from some amounts of transferred charge that is contributing to the modified ORR process and, hence, is not contributing to the actual salt removal.
The CDI evaluation of FW200 and FW200/TiO2 in de-aerated 5 mM NaCl (i.e., the typical electrolyte used for most CDI work) is shown in Fig. 5. At first, FW200 displays a small inverse peak before starting the adsorption process again (Fig. 5A), but after 15 cycles, the inverse peak vanished (Fig. 5B). The decrease of the inverse peak may be linked to progressing carbon oxidation at the positive electrode. In contrast, FW200/TiO2 exhibits rather constant inverse peaks (Fig. 5A and B). The salt adsorption performance of FW200 and FW200/TiO2 is shown in Fig. 5C. The SAC of FW200 and FW200/TiO2 starts at 5.5 mg g−1 and slightly decays before stabilizing around 2 mg g−1 after 20 CDI cycles. Thus, the SAC performance in de-aerated aqueous solution is rather similar for FW200 and FW200/TiO2, leading to an unfavorable decay in performance of more than 50% of the initial value over just 10 cycles. Partially inverted CDI also leads to an unfavorably low charge efficiency of 20–30% after 20 CDI cycles (Fig. 5D). Considering these performance values, FW200, with or without titania decoration, is unfavorable for use in de-aerated aqueous solution. Seemingly, the majority phase (FW200) and its associated surface functionalities dominate the electrochemical performance in de-aerated solutions, exhibiting low charge efficiency and fast decay of SAC performance.
 | ||
| Fig. 5 CDI performance of FW200 and FW200/TiO2 in de-aerated 5 mM NaCl with 22 mL min−1 of flow rate. The applied cell voltage is 1.2 V during charging and 0 V during discharging. (A and B) Conductivity at 5th and 15th cycle. (C) Salt adsorption capacity over 30 cycles. (D) Charge efficiency corresponds to the data shown in panel (C). | ||
For comparison, we synthesized activated carbon/titania hybrids by using the same method as FW200/TiO2 and tested the CDI performance in oxygen saturated 5 mM NaCl. We chose commercial YP-80F (Kuraray) with a BET surface area of 2347 m2 g−1, which is characterized by a large inner porosity.43 As shown in Fig. S1 (ESI†), pristine activated carbon exhibits a SAC of 9 mg g−1 in the first cycle and a drastic decrease to nearly zero after 15 cycles. This fading in CDI performance of pure activated carbon is related to the oxidization of the anode, comparable to what we have shown for FW200. Once titania is coated on the activated carbon surface, the prolongation of CDI performance in oxygen saturated solution can be seen. The activated carbon/titania hybrid shows a very high initial SAC of 18 mg g−1 in the first cycle, but decreases to a rather constant value of ca. 2 mg g−1 after 60 cycles. Thus, while titania nanodecoration of activated carbon seemingly improves the performance and CDI stability, the improvement is inferior to what is seen for FW200/TiO2, where the majority of surface area is associated with outer surface.
We further studied the catalytic activity (ORR) of titania decoration on FW200 by use of a rotating ring electrode (RDE). As shown in Fig. 6A, linear sweep voltammograms at 10 mV s−1 and 0 rpm of FW200 and FW200/TiO2 exhibit an ORR onset potential of −0.086 V and 0.049 V vs. Ag/AgCl, respectively. The higher onset potential of FW200/TiO2 implies that titania catalyzes ORR. To obtain further information about ORR including the influence of mass diffusion on ORR, RDE measurements with various rotating speeds (200–3200 rpm) were carried out (Fig. 6A and B). For FW200 and FW200/TiO2, the diffusion current (iL) is increased when increasing the rotation speed due to the reduction of diffusion length. The measured current at −0.3 V vs. Ag/AgCl of FW200 and FW200/TiO2 shows a rather linear correlation with the square root of the rotation speed. The resulting slope of the K–L plot (Fig. 6D) according to eqn (2) and (3) reflects the mechanism by the estimation of n. As shown in Fig. 6D, FW200 presents 1.4 electron transfers, while FW200/TiO2 displays a four electron transfer. As identified in eqn (4)–(6), the four electron transfer (eqn (4)) is favorable for CDI longevity because only hydroxyl ions are formed as reactants. However, in our case, the four electron pathway is not preferred when carbon has a high amount of oxygen functional groups (9 mass%). Earlier work has shown that oxygen functional groups including carbonyl, carboxyl, and hydroxyl dangling bonds on carbon nanotubes exhibit a two electron pathway at a potential of −0.6 V vs. Ag/AgCl with an onset potential of ca. −0.1 V vs. Ag/AgCl.44 Therefore, the results shown in the inserted table in Fig. 6D are in good alignment with previous work, since FW200 exhibits 1.4 electron transfers leading to hydrogen peroxide formation (eqn (5)).
| O2 + 2H2O + 4e− → 4OH− | (4) |
| O2 + H2O + 2e− → HO2− + OH− | (5) |
| HO2− + H2O + 2e− → 3OH− | (6) |
 | ||
| Fig. 6 (A) Linear sweep voltammograms (LSV) at 10 mV s−1 of FW200 and FW200/TiO2 in oxygen saturated 1 M NaCl at 0 rpm, (B and C) LSV at 10 mV s−1 of FW200 (B) and FW200/TiO2 (C) in O2 saturated 1 M NaCl with various rotation speeds, and (D) Koutecky–Levich (K–L) plot at −0.3 V vs. Ag/AgCl. | ||
We expect initial H2O2 is further reduced on FW200/TiO2. The latter has the catalytic ability to modify the oxygen reduction reaction, reducing H2O2 to hydroxide (eqn (6)),45 as enabled by the four electron transfer in FW200/TiO2 (inserted table in Fig. 6D). The observed CDI behavior is explained by a transition of the two electron transfer reaction for FW200 that leads to peroxide formation, to a four electron transfer reaction for FW200/TiO2, thereby further reducing peroxide evolution and effectively preventing the oxidative carbon degradation. This explanation requires more comprehensive follow-up work to further elucidate the precise mechanisms behind this intriguing performance stability. In absence of excess oxygen, in de-aerated water, ORR does not occur and the carbon degeneration per surface functional groups seemingly dominates the decay of the CDI performance.
Conclusions
The titania decoration of carbon proved to be an excellent and facile approach to overcome the limited CDI performance stability in oxygen saturated saline solutions. Starting with a promising SAC value of 10 mg g−1, carbon black shows a negligible salt removal capacity of 2 mg g−1 after just 20 CDI cycles. Titania-decorated carbon black showed a stable performance of 7 ± 1 mg g−1 over a remarkable duration of 100 CDI cycles, being by far the most stable CDI system not using membranes in oxygen saturated media. Future work beyond this proof-of-concept study will have to establish the exact mechanism behind this intriguing performance stability and find the optimized amount of titania loading, while extending the scope to other carbon materials.Acknowledgements
This work was carried out within the framework of the Franco-German EEIGM Master's Program in Materials Science at Saarland University. S. P. acknowledges financial support by the German Academic Exchange Service (DAAD award number 91579066). The authors thank Prof. Eduard Arzt (INM) for his continuing support.References
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 CAS.
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS.
- J. W. Blair and G. W. Murphy, in Saline Water Conversion, American Chemical Society, 1960, vol. 27, ch. 20, pp. 206–223 Search PubMed.
- Y. Liu, C. Y. Nie, X. J. Liu, X. T. Xu, Z. Sun and L. K. Pan, RSC Adv., 2015, 5, 15205–15225 RSC.
- T. Kim, J. E. Dykstra, S. Porada, A. van der Wal, J. Yoon and P. M. Biesheuvel, J. Colloid Interface Sci., 2015, 446, 317–326 CrossRef CAS PubMed.
- X. Xu, L. Pan, Y. Liu, T. Lu, Z. Sun and D. H. C. Chua, Sci. Rep., 2015, 5, 8458 CrossRef CAS PubMed.
- X. Xu, Z. Sun, D. H. C. Chua and L. Pan, Sci. Rep., 2015, 5, 11225 CrossRef CAS PubMed.
- X. Gao, A. Omosebi, J. Landon and K. L. Liu, Energy Environ. Sci., 2015, 8, 897–909 CAS.
- I. Cohen, E. Avraham, Y. Bouhadana, A. Soffer and D. Aurbach, Electrochim. Acta, 2013, 106, 91–100 CrossRef CAS.
- A. Omosebi, X. Gao, J. Landon and K. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12640–12649 CAS.
- X. Gao, A. Omosebi, J. Landon and K. Liu, J. Electrochem. Soc., 2014, 161, E159–E166 CrossRef.
- A. Omosebi, X. Gao, J. Rentschler, J. Landon and K. Liu, J. Colloid Interface Sci., 2015, 446, 345–351 CrossRef PubMed.
- X. Gao, A. Omosebi, J. Landon and K. Liu, Environ. Sci. Technol., 2015, 49, 10920–10926 CrossRef CAS PubMed.
- X. Gao, S. Porada, A. Omosebi, K.-L. Liu, P. M. Biesheuvel and J. Landon, Water Res., 2016, 92, 275–282 CrossRef CAS PubMed.
- P. B. Peters, R. van Roij, M. Z. Bazant and P. M. Biesheuvel, 2016, ArXiv, 1512.04261.
- D. He, C. E. Wong, W. Tang, P. Kovalsky and T. D. Waite, Environ. Sci. Technol. Lett., 2016, 3, 222–226 CrossRef CAS.
- T. Kim, J. Yu, C. Kim and J. Yoon, J. Electroanal. Chem., 2016, 776, 101–104 CrossRef CAS.
- J. Yang, L. Zou, H. Song and Z. Hao, Desalination, 2011, 276, 199–206 CrossRef CAS.
- M. T. Z. Myint and J. Dutta, Desalination, 2012, 305, 24–30 CrossRef CAS.
- M.-W. Ryoo, J. H. Kim and G. Seo, J. Colloid Interface Sci., 2003, 264, 414–419 CrossRef CAS PubMed.
- J.-W. Lee, H.-I. Kim, H.-J. Kim and S.-G. Park, Appl. Chem. Eng., 2010, 21, 265–271 CAS.
- C. Kim, J. Lee, S. Kim and J. Yoon, Desalination, 2014, 342, 70–74 CrossRef CAS.
- J. J. Lado, R. E. Pérez-Roa, J. J. Wouters, M. Isabel Tejedor-Tejedor and M. A. Anderson, Sep. Purif. Technol., 2014, 133, 236–245 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- G. Y. Gor, M. Thommes, K. A. Cychosz and A. V. Neimark, Carbon, 2012, 50, 1583–1590 CrossRef CAS.
- M. Aslan, D. Weingarth, P. Herbeck-Engel, I. Grobelsek and V. Presser, J. Power Sources, 2015, 279, 323–333 CrossRef CAS.
- X. Xu, M. Wang, Y. Liu, T. Lu and L. Pan, J. Mater. Chem. A, 2016, 4, 5467–5473 CAS.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS PubMed.
- G. W. Hung and R. H. Dinius, J. Chem. Eng. Data, 1972, 17, 449–451 CrossRef CAS.
- J. Kestin, H. E. Khalifa and R. J. Correia, J. Phys. Chem. Ref. Data, 1981, 10, 71–88 CrossRef CAS.
- J. E. Sherwood, F. Stagnitti, M. J. Kokkinn and W. D. Williams, Limnol. Oceanogr., 1991, 36, 235–250 CrossRef CAS.
- M. Aslan, M. Zeiger, N. Jäckel, I. Grobelsek, D. Weingarth and V. Presser, J. Phys.: Condens. Matter, 2016, 28, 114003 CrossRef CAS PubMed.
- B. Gao, G. Z. Chen and G. Li Puma, Appl. Catal., B, 2009, 89, 503–509 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 075414 CrossRef.
- S. Fleischmann, N. Jäckel, M. Zeiger, B. Krüner, I. Grobelsek, P. Formanek, S. Choudhury, D. Weingarth and V. Presser, Chem. Mater., 2016, 28, 2802–2813 CrossRef CAS.
- M. Zeiger, N. Jäckel, V. N. Mochalin and V. Presser, J. Mater. Chem. A, 2016, 4, 3172–3196 CAS.
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS PubMed.
- Y. Bouhadana, E. Avraham, M. Noked, M. Ben-Tzion, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2011, 115, 16567–16573 CAS.
- X. Sun, P. Song, Y. Zhang, C. Liu, W. Xu and W. Xing, Sci. Rep., 2013, 3, 2505 Search PubMed.
- J. Zhang and L. Dai, ACS Catal., 2015, 5, 7244–7253 CrossRef CAS.
- R. Zhao, P. M. Biesheuvel, H. Miedema, H. Bruning and A. van der Wal, J. Phys. Chem. Lett., 2009, 1, 205–210 CrossRef.
- S. Porada, F. Schipper, M. Aslan, M. Antonietti, V. Presser and T.-P. Fellinger, ChemSusChem, 2015, 8, 1867–1874 CrossRef CAS PubMed.
- N. Jäckel, M. Rodner, A. Schreiber, J. Jeongwook, M. Zeiger, M. Aslan, D. Weingarth and V. Presser, J. Power Sources, 2016, 326, 660–671 CrossRef.
- R.-S. Zhong, Y.-H. Qin, D.-F. Niu, J.-W. Tian, X.-S. Zhang, X.-G. Zhou, S.-G. Sun and W.-K. Yuan, J. Power Sources, 2013, 225, 192–199 CrossRef CAS.
- J. Yu, Z. Liu, L. Zhai, T. Huang and J. Han, Int. J. Hydrogen Energy, 2016, 41, 3436–3445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22800c |
| This journal is © The Royal Society of Chemistry 2016 |