
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of the release of functional payloads from redox-responsive nanocapsules†
Shuai
Jiang
ab,
Katharina
Landfester
a and
Daniel
Crespy
*ac
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: crespy@mpip-mainz.mpg.de
bInstitute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
cDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand
First published on 25th October 2016
Abstract
Stimuli-responsive nanocontainers allow for storage and controlled release of functional payloads in response to external stimuli. However, release profiles in which no leakage occurs without stimuli and complete release is achieved upon activation, are still a challenge. Herein, we conducted a systematic study of the factors contributing to the final release profiles of payloads from nanocapsules that are the diffusion of payloads due to the porosity of the shell and the release of payloads after activation with an external stimulus. This study provides fundamental information for designing nanocarriers that are able to effectively encapsulate and control release of payloads in response to the change in their surrounding environment.
Introduction
Stimuli-responsive materials can sense changes in the surrounding environment and respond actively through physical and chemical changes.1 Nanocarriers with stimuli-responsive properties, allowing for storage and controlled release of payloads in response to specific stimuli, are playing an increasingly important part in the fields of drug delivery2–4 and corrosion protection.5–7 On-demand drug delivery is thereby becoming feasible through the design of stimuli-responsive nanocarriers that can recognize their microenvironment and react in a dynamic way, mimicking the responsiveness of living organisms.2,8Herein, we design redox-responsive nanocapsules, i.e. nanocarriers with a core–shell structure. The core of capsules possesses a high encapsulation capacity for functional payloads. The capsule shell provides a protection as well as enables controlled release of payloads in response to the change of the redox potential. A comparative study of the main factors, shell porosity and responsive gates, contributing to the final release profiles of the payloads is presented.
Owing to the impressive progress in biomedical and materials science, a broad range of nanocarriers have been developed with diverse architectures and chemistries.8 These nanocarriers include liposomes, micelles, dendrimers, nanoparticles, nanocapsules, and nanofibers.8–12 Nanocapsules, due to their core–shell structure, exhibit unique advantages for storage and controlled release of functional payloads. Firstly, processes such as the emulsion–solvent evaporation technique13 and miniemulsion polymerization9,14–16 allow the formation of nanocapsules with a liquid core that can be hydrophobic (oily) or hydrophilic (in many cases aqueous). Thereby either hydrophobic or hydrophilic payloads can be encapsulated in the liquid core. Secondly, the shell of nanocapsules serves as a smart barrier which provides a protection of the payloads from the surrounding environment as well as an enhanced control of the release of the payloads. These features displayed by stimuli-responsive nanocapsules are also present in nature. Indeed the cell membrane protects enzymes and organelles in the interior of the cell from the extracellular environment and controls the secretion of hormones into the surrounding environment.17 Moreover, both the core and the shell of nanocapsules can be utilized for the loading of multiple agents. Functional payloads can either be physically entrapped in the core of nanocapsules15 or be placed on the shell's surface by physio- or chemisorption18 or chemical bonding.19 Selective release of payloads can thereby be carried out by increasing the permeability of the capsule shell15 or by desorption from the shell.20 The selective release of one payload upon several stimuli16 or several payloads upon several stimuli can be also realized.19
The implementation of such stimuli-responsive devices requires the use of materials that in response to a specific stimulus, undergo a protonation, a hydrolytic cleavage or a supramolecular conformational change.8 Materials with redox-responsive properties have recently received tremendous attention for drug delivery8,21 and corrosion protection.5 One route to synthesize redox-responsive materials is using ferrocene as redox-responsive units.22 Ferrocene experiences a transition from hydrophobic to hydrophilic upon oxidation. Based on this ferrocene/ferrocenium pair, redox-responsive patchy nanocapsules,16,23 microcapsules fabricated by layer-by-layer method,21 and crosslinked poly(ionic liquid)s hydrogel24 were developed for controlled release of payloads upon oxidation. Approaches based on disulfide,25 diselenide,26 host–guest polymers,27 polyaniline,15 thiol–gold interaction,19 quinone derivatives,28 supramolecular chemistry,29 as well as oxidation of sulfides30 and selenium-containing polymers31 have also been developed for the fabrication of redox-responsive nanocarriers.32
Interconversion between thiols and disulfides is a key step in many biological processes and plays an important role in the stability and rigidity of native proteins in living cells.25 Therefore, the disulfide linkage is an ideal candidate for designing redox-responsive nanocarriers for biomedical applications. The synthesis and subsequent self-assembly of amphiphilic block copolymers into micelles have been the focus of considerable research. In general, such redox-responsiveness can readily be achieved by incorporating disulfide moieties into the block copolymer micelles, either between the hydrophobic and hydrophilic blocks,33–36 or in the crosslinkers.37,38 These micelles can be disassembled followed by specific intracellular release of hydrophobic drugs. Capsules with a core–shell structure represent another field where disulfide linkages were used. Redox-responsive silica39 and polymer nanocapsules40 were synthesized by incorporating disulfide bonds in the shell, through which the shell permeability can be tuned by reduction and oxidation. Disulfide linkages can be incorporated in the polymer backbone, at the side chains of polymer, or in the crosslinker molecule. Functional payloads are then trapped in the core of nanocontainers or grafted on the side chains by cleavable disulfide linkage.20 Other redox-responsive systems based on disulfide crosslinkers include microcapsules engineered by the layer-by-layer method,41,42 silica/polymer double-walled hybrid nanotubes,43 hydrogels,8,44,45 and electrospun nanofibers.10
In this study, we synthesize nanocapsules by using materials with different porosity and containing different redox-responsive units. A systematic investigation of the main factors contributing to the final release profiles was conducted through studying comparatively the redox-responsive release of functional payloads from nanocapsules. Disulfide and tetrasulfide bonds were introduced as redox-responsive units in the capsule shell composed of silica or polymer. To verify the applicability of our nanostructured systems, the corrosion inhibitor 2-mercaptobenzothiazole (MBT)46 and the anti-inflammatory drug indomethacin (IMC) were selected as functional payloads encapsulated in the core. In particular, we investigated here: (1) the influence of shell porosity on the release profiles of payloads; (2) the influence of the redox-responsive gates on the selectivity of release; and (3) the effect of the size of payload molecules on the selectivity of release.
Experimental
Materials
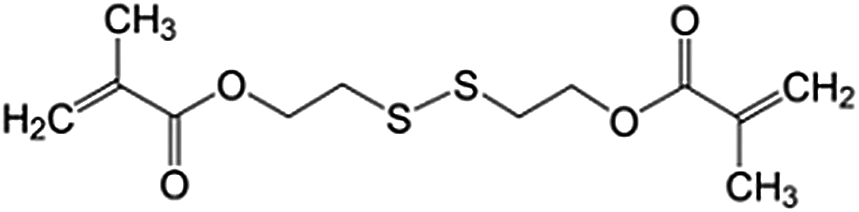
Tetraethoxysilane (TEOS, Alfa Aesar, 98%), bis[3-(triethoxysilyl)propyl] tetrasulfide (TESPT, Sigma Aldrich, 90%), bis(triethoxysilylpropyl) disulfide (TESPD, Gelest, 90%), bis(2-methacryloyl)oxyethyl disulfide (DSDMA, Sigma Aldrich), methyl methacrylate (MMA, Acros, 99%), 2,2′-azobis(2-methylbutyronitrile) (AMBN, Wako Chemicals), hexadecane (HD, Sigma Aldrich, 99%), m-xylene (Acros Organics, ≥99%), 2-mercaptobenzothiazole (MBT, Sigma Aldrich, 97%), indomethacin (IMC, Sigma Aldrich, ≥99%), cetyltrimethylammonium chloride (CTMA-Cl, Acros Organics, 99%), sodium dodecyl sulfate (SDS, Alfa Aesar, 99%), 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS, Sigma Aldrich, 97%), and tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl, Alfa Aesar, ≥98%) were used as received. Demineralized water was used through all the experiments if not specifically mentioned.Encapsulation of functional payloads in redox-responsive silica nanocapsules
Redox-responsive silica nanocapsules loaded with MBT or IMC were synthesized by miniemulsion polymerization.39 Alkoxysilane precursors were first mixed with 125 mg of HD, 1 g of m-xylene, and 25 mg of MBT or IMC, and then added to 30 mL of a 0.77 mg mL−1 aqueous solution of CTMA-Cl. For the synthesis of silica capsules containing tetrasulfide bonds, 1.5 g of TEOS and 0.5 g of TESPT were used. In the case of silica capsules containing disulfide bonds, the same molar amount of TESPD (0.44 g) was used instead of TESPT. After pre-emulsification at 1000 rpm for 1 h at room temperature, the emulsion was sonicated under ice cooling for 180 s at 70% amplitude in a pulse regime (30 s sonication, 10 s pause) using a Branson 450 W sonifier and a 1/2′′ tip. The resulting miniemulsions were stirred at 1000 rpm for 12 h at room temperature to obtain the silica nanocapsules.Encapsulation of functional payloads in redox-responsive polymer nanocapsules
Monomers (2.0 g of DSDMA or a mixture of 0.2 g of DSDMA and 1.8 g of MMA) were first mixed with 100 mg of AMBN, 2 g of HD, 2 g of m-xylene, and 25 mg of MBT, and then added to 24 mL of a 0.5 mg mL−1 aqueous solution of SDS. After pre-emulsification at 1000 rpm for 1 h at room temperature, the emulsion was sonicated under ice cooling for 180 s at 70% amplitude in a pulse regime (30 s sonication, 10 s pause) using a Branson 450 W sonifier and a 1/2′′ tip. The resulting miniemulsions were stirred (1000 rpm) at 72 °C for 12 h to obtain the polymer nanocapsules. The conversion of monomers was determined by measuring the dispersion of nanocapsules in D2O with 1H NMR spectroscopy using DSS as internal standard.Controlled release of payloads from nanocapsules
To study the redox-responsive release of payloads from the nanocapsules, 0.1 mL of the nanocapsule dispersions and 0.9 mL of aqueous dialysis medium were placed into a dialysis bag (MWCO 14000). Afterwards, the dialysis bag was immersed into 9 mL of an aqueous dialysis medium. In the case of silica nanocapsules containing tetrasulfide bonds, an aqueous solution of 0.70 mmol L−1 TCEP·HCl (corresponding to molar ratio of TCEP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TESPT = 3:1) was used as the dialysis medium whereas an aqueous solution of 0.23 mmol L−1 TCEP·HCl (corresponding to molar ratio of TCEP:TESPD = 1:1) was used for the silica nanocapsules containing disulfide bonds. In the case of polymer nanocapsules composed of 10% and 100% DSDMA in the shell, 0.066 or 0.66 mmol L−1 TCEP·HCl was used as the dialysis medium. For the control samples, demineralized water was used in all release experiments. After given time intervals, 0.4 mL of the solution outside the dialysis bag was removed for UV-Vis measurements and replaced by an equal amount of reducing agent/water solution to keep the volume constant. The concentration of released payloads was determined by UV-Vis spectroscopy.
:TESPT = 3:1) was used as the dialysis medium whereas an aqueous solution of 0.23 mmol L−1 TCEP·HCl (corresponding to molar ratio of TCEP:TESPD = 1:1) was used for the silica nanocapsules containing disulfide bonds. In the case of polymer nanocapsules composed of 10% and 100% DSDMA in the shell, 0.066 or 0.66 mmol L−1 TCEP·HCl was used as the dialysis medium. For the control samples, demineralized water was used in all release experiments. After given time intervals, 0.4 mL of the solution outside the dialysis bag was removed for UV-Vis measurements and replaced by an equal amount of reducing agent/water solution to keep the volume constant. The concentration of released payloads was determined by UV-Vis spectroscopy.
Characterization
The intensity-average hydrodynamic diameter of nanocapsules was evaluated by dynamic light scattering (DLS) using a Nicomp particle sizer (Model 380, PSS, Santa Barbara, CA) at a fixed scattering angle of 90°. The shell thickness of the nanocapsules was measured by counting at least 100 capsules from TEM micrographs. The morphologies of nanocapsules were examined with a Gemini 1530 (Carl Zeiss AG, Oberkochem, Germany) scanning electron microscope operating at 0.35 kV and a Jeol 1400 (Jeol Ltd, Tokyo, Japan) transmission electron microscope operating at an accelerating voltage of 120 kV. Scanning and transmission electron microscopy (SEM and TEM) specimens were prepared by evaporating one drop of diluted nanocapsule dispersion on silicon wafers and 400-mesh carbon layer-coated copper grids, respectively. The concentration of released payloads in the dialysis medium was measured with a Perkin Elmer Lambda 25 UV-Vis spectroscopy. The surface area of nanocapsules before and after reduction was determined from nitrogen adsorption–desorption experiments that were carried out on a Quantachrome Autosorb-1 analyzer (Boynton Beach, FL) at 77.3 K. The specimens were obtained by first adding a calculated amount of TCEP·HCl in the dispersion of nanocapsules and stirring for 12 h for the reduction of disulfide and tetrasulfide linkages. Then the capsule dispersions were dialyzed in demineralized water for 3 days. The dialyzed dispersion was then freeze-dried for 48 h and degassed at 70 °C for at least 12 h under high vacuum before measurements. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) equation based on data points obtained from 0 < P/P0 < 0.25. 1H NMR spectra were measured at room temperature on a console Avance 300.Results and discussion
Encapsulation of functional payloads in redox-responsive nanocapsules
Nanocapsules with core–shell morphology were synthesized in miniemulsion (see Fig. 1a). The functional payloads were first dissolved in an organic phase (HD/m-xylene/monomers) forming the droplets and then were encapsulated in situ in the core of the nanocapsules. HD was added as a hydrophobe to stabilize the emulsion against Ostwald ripening. m-Xylene served as solvent for the payloads MBT and IMC. The silica precursors, i.e. tetraethoxysilane and an alkoxysilane containing disulfide bonds or tetrasulfide bonds, were used to build the silica shell of capsules which are denoted as NCSi2S and NCSi4S (see Table 1). Through introducing these redox-responsive units, shell permeability can be tuned by applying a reducing and oxidizing trigger.39 The monomers MMA and DSDMA, the latter containing disulfide bonds, were used to construct the redox-responsive polymer nanocapsule denoted NCP2S in the text (see Table 1). Well-defined core–shell morphologies were identified by SEM and TEM in all cases (see Fig. 1b and c). The formation of core–shell morphology in silica nanocapsules is due to the hydrolysis and condensation of alkoxysilane precursors at the interface between miniemulsion droplets and the water phase. In the case of polymer nanocapsules, the core–shell morphology resulted from the precipitation of the polymer chains at the interface of the droplets due to the poor solubility of the formed polymer in both the non-solvents present in the dispersed phase and in the water phase.15 The conversion of DSDMA in miniemulsion polymerization was determined to be 98% by comparing the signal of DSDMA in 1H NMR spectra before and after the polymerization reaction and using DSS as an internal standard compound (see ESI Fig. S1†).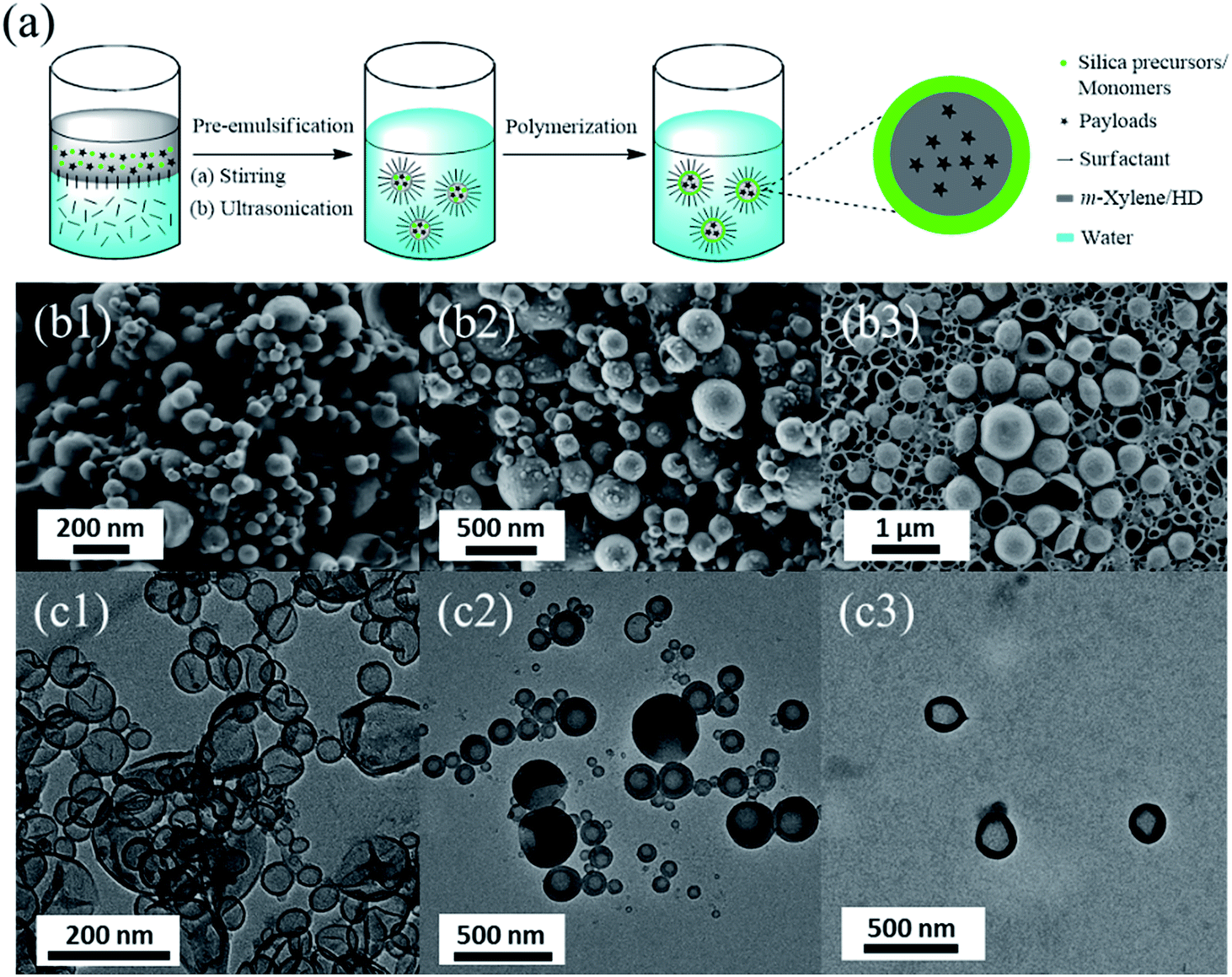 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of nanocapsules by miniemulsion polymerization. SEM (b1–3) and TEM micrographs (c1–3) of silica nanocapsules NCSi4S (b1 and c1) and NCSi2S (b2 and c2), and of the polymer nanocapsules NCP2S (b3 and c3). | ||


Controlled release of MBT from silica nanocapsules containing tetrasulfide and disulfide bonds
The redox-responsive property of the nanocapsules was investigated by studying the release profiles of their payloads. A corrosion inhibitor MBT and an anti-inflammatory drug IMC were encapsulated as functional payloads. Since the release study was performed in aqueous medium, hydrophilic reducing agents dithiothreitol (DTT) and TCEP·HCl were tried for the reduction of disulfide bonds. DTT is effective and widely used for the cleavage of disulfide bonds in biological system.47 However, the reducing power of DTT is limited to pH values above 7, since only the negatively charged thiolate form –S− is reactive while the protonated thiol form –SH is not.48 Also because the UV absorbance of oxidized DTT overlaps the absorbance of MBT, the quantification of released MBT is thereby difficult. Therefore, TCEP, an highly effective reducing agent for the reduction of disulfide bonds, was adopted for the subsequent study.49 The release of payloads was then followed by using UV-Vis spectroscopy.Firstly, a comparative study aiming to understand the effect of redox-responsive gates on the release profiles was conducted by studying the release of MBT from silica nanocapsules containing tetrasulfide or disulfide bonds. It has to be noted that the release profiles of NCSi4S in Fig. 2b (molar ratio of TCEP:tetrasulfide bonds = 0:1 and 3:1) have been reported in our previous study and used here as a control sample.50 When no reducing trigger was applied, NCSi4S showed a burst release of ∼55% MBT (compared to the initial amount of MBT in the capsules) in the first hour (see Fig. 2b). This non-specific release, i.e. release that occurs independently on the redox conditions, is due to the intrinsic porosity of the silica shell. Part of the encapsulated MBT molecules could successfully diffuse through the interconnected pores of the silica shell into the dialysis medium. The release of MBT was dramatically accelerated in an aqueous medium with 0.70 mmol L−1 TCEP·HCl. This accelerated release was attributed to the reduction of tetrasulfide linkages, which lead to an increase of the permeability of silica shell.39 When the release equilibrium was reached, almost 100% release of MBT from NCSi4S was observed. Comparatively, a non-specific release of ∼45% MBT was observed in 8 h in the case of NCSi2S. This relatively lower and slower release of MBT can be attributed to the thicker shell of NCSi2S. A shell thickness of 13 nm was observed in the TEM micrograph of NCSi2S compared to a shell thickness of 7 nm in the capsules NCSi4S (see Table 1). The thicker shell results in longer pathways for the diffusion of payloads, thereby lead to a slower release profile. When the release equilibrium was reached, 70% of MBT released from NCSi2S in a slower way (in 8 h), which can be explained by the smaller gate opened by the reduction of disulfide bonds compared with tetrasulfide bonds.
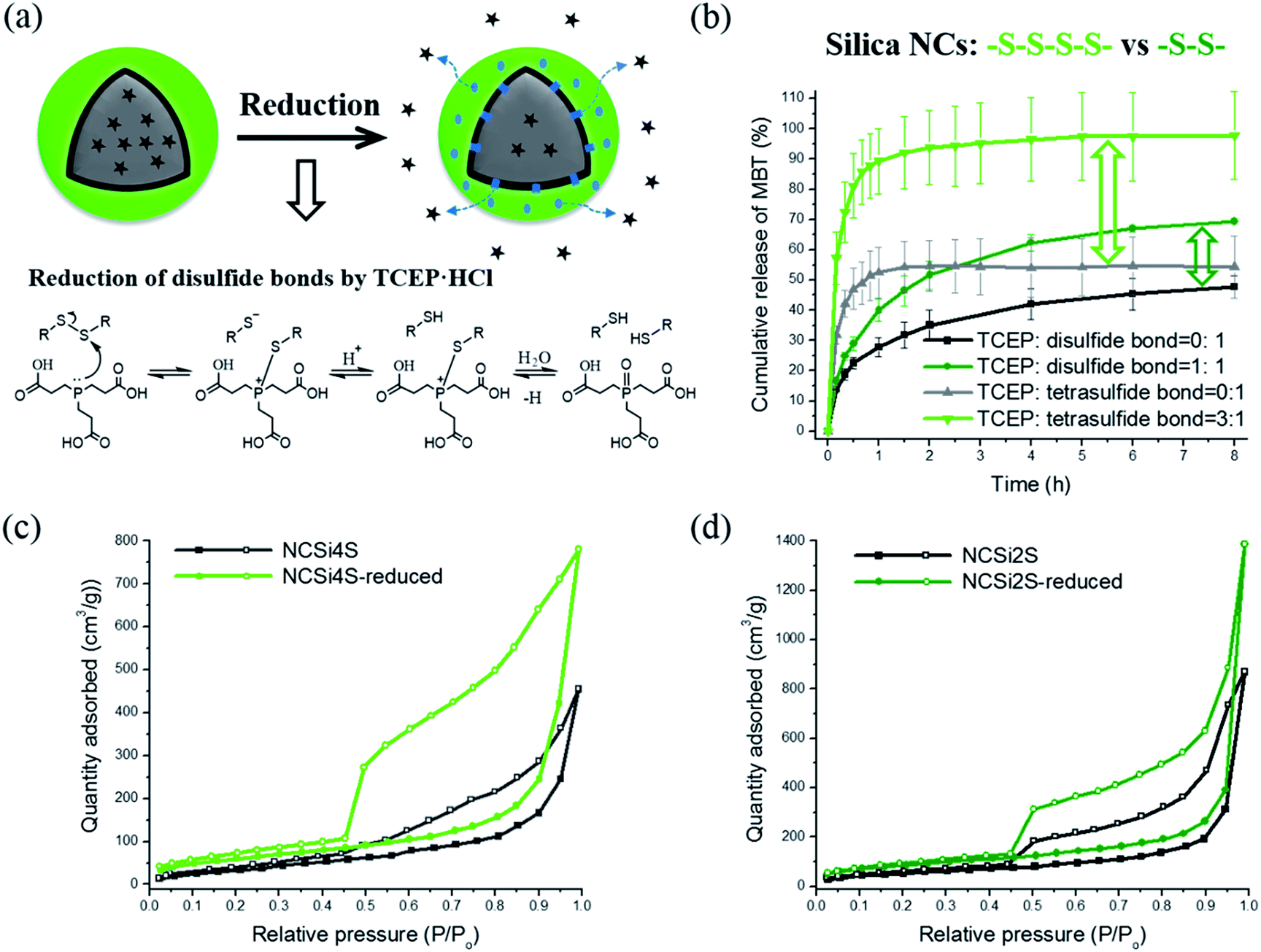 | ||
| Fig. 2 (a) Scheme depicting the mechanism of redox-responsive release of payloads from the nanocapsules; (b) release profiles of MBT from NCSi4S and NCSi2S; nitrogen adsorption–desorption isotherms of dry silica nanocapsules (c) NCSi4S and (d) NCSi2S before and after reduction. | ||
In real applications for corrosion protection, nanocontainers are usually incorporated in the protective coatings on metal substrates. The coating matrix could prevent the unwanted leakage of corrosion inhibitor before corrosion occurs. In a parallel study, redox-responsive silica nanocapsules were embedded in polyvinyl alcohol nanofibers by using colloid-electrospinning.50 The polymer fiber matrix was able to effectively inhibit the leakage of MBT from the capsules NCSi4S (the release equilibrium decreased from 55% to 20%). Moreover, the release of MBT from the nanofibers was much slower than the release from the nanocapsules. For the ratio TCEP:TESPT = 0.3:1, the duration for the release of 80% of the initial quantity of the payload is 23 times higher than that for nanocapsules. This hindrance of burst release is attributed to the barrier effect of the polymer matrix, yielding longer pathways for the molecules involved in the chemical stimulus and the molecules of the payloads.
Furthermore, the influence of pH on the reduction-induced release was studied by monitoring the release profiles of MBT from NCSi4S with TCEP concentration of 0.70 mmol L−1 at pH = 2, 7, and 12. It was found that, the release rate and equilibrium concentration of MBT from NCSi4S under reducing environment increased as the pH value of the release medium increased (see Fig. 3). Since the pKa of MBT is 6.93,51 acceleration of the release of MBT was observed in the cases of pH = 7 and 12 in comparison with the case of pH = 2 due to the deprotonation of the thiol groups at high pH.
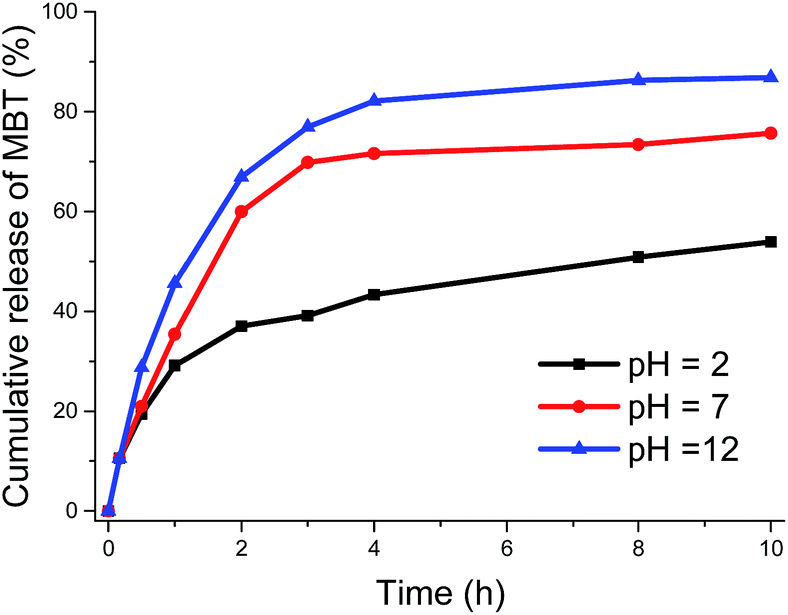 | ||
| Fig. 3 Release profiles of MBT from NCSi4S triggered by TCEP·HCl at pH = 2, 7, and 12. | ||
To investigate the change of capsule shell before and after applying the reducing trigger, nitrogen adsorption–desorption experiments were conducted. From Fig. 2c and d, we can clearly see the change of nitrogen adsorption–desorption isotherms of silica nanocapsules before and after reduction. When the reducing trigger was applied, an increase of 58% and 57% surface area was observed in the case of NCSi4S and NCSi2S, respectively, due to the cleavage of redox-responsive linkages in the shell. SEM micrographs in Fig. S2† indicated that the silica nanocapsules were kept integrity after the drying process for BET measurements.
Comparative study of the release profiles of MBT from silica and polymer nanocapsules containing disulfide bonds
The porosity of the silica shell plays a main part in the burst release of payloads as discussed above. To further investigate the effect of shell porosity on the release profile of payloads, polymers containing disulfide bonds were used to build the shell of nanocapsules instead of silica. During the polymerization process, the growing polymer chain phase separated and was not dissolved in the solvent any more. It was therefore deposited at the oil/water interface as a dense polymer wall with a relatively low permeability, as indicated by a low BET surface area of NCP2S in Table 1. Compared with the nonspecific release of ∼45% MBT from NCSi2S, the burst release of MBT was significantly suppressed in the case of polymer nanocapsules with a nonspecific release of ∼15% MBT (see Fig. 4a). This observation is consistent with the BET surface of the capsules shown in Table 1. The porous NCSi2S possess a BET surface of 198 m2 g−1 compared with a value of 19 m2 g−1 of the NCP2S. When the reducing trigger was applied, the release of MBT from NCP2S increased to ∼40% with an improvement of 25% which is very similar with the increase in the case of NCSi2S. This increase of release upon reduction can be defined as selectivity of redox-responsive release, which represents the gap between the release equilibrium with and without reducing trigger. The similar selectivity of redox-responsive release in both cases of NCSi2S and NCP2S indicated that the specific release of MBT was controlled by the redox-responsive gates in the shell. Polymer nanocapsules with the shell consisting of 10 wt% DSDMA and 90 wt% MMA were also synthesized and compared with the case of NCP2S whose shell is composed of 100% DSDMA (see Fig. S3†). A slight decrease of both nonspecific release and specific release under reduction was observed in the case of 10 wt% of DSDMA contained. | ||
| Fig. 4 (a) Release profiles of MBT from NCSi2S and NCP2S; (b) nitrogen adsorption–desorption isotherms of dry polymer nanocapsules NCP2S before and after reduction. | ||
Comparative study of the release of IMC from silica nanocapsules containing tetrasulfide and disulfide bonds
To further investigate the influence of redox-responsive linkages on the selectivity of the release, the anti-inflammatory drug IMC with a larger molecular weight (358 g mol−1) than the one of MBT (167 g mol−1) was entrapped in silica nanocapsules. From the discussion above, we can conclude that two main aspects contribute to the final release profiles of payloads that are (a) the diffusion due to the porosity of the shell and (b) the specific release from the gate opened by reduction. From the release curve of IMC in Fig. 5, a non-specific release of ∼10% IMC (compared to the initial amount of IMC in the nanocapsules) was observed in silica nanocapsules NCSi2S and NCSi4S, which can be attributed to the diffusion due to the porosity of silica shell. Compared with the nonspecific release of 45–55% MBT from silica nanocapsules, the nonspecific release of IMC was suppressed due to its larger size. No acceleration of IMC release was observed when reducing trigger was applied. During the reduction process, only the redox-responsive linkages were cleaved while the rigid silica network of the shell was kept (see Fig. S4†). Due to the dimension limitation of opened disulfide and tetrasulfide gates in the shell, the transportation of encapsulated IMC molecules was still hindered. These results provide useful information for the development of nanocarriers that can effectively encapsulate and control release of functional molecules through designing the stimuli-responsive gates. | ||
| Fig. 5 Release profiles of IMC from NCSi2S and NCSi4S. | ||
Remarkably, nanocapsules constructed by both silica and polymer were kept intact after triggered release and their core–shell morphology can still be identified by TEM (see Fig. S4†). In fact, only redox-responsive units, disulfide or tetrasulfide bridges, were opened under reduction. The whole silica or polymeric networks can be kept which is not the case of stimuli-responsive micelles that disassembled under the triggers.
Conclusions
Controlled release of functional payloads in response to the change of redox-potential was achieved through the design of nanocapsules with different compositions. Reduction cleavable disulfide or tetrasulfide linkages were introduced in the nanocapsules shell, through which the shell permeability can be controlled upon redox stimuli. The porosity of the shell and the activation of capsule shell with an external stimulus were found to be main factors contributing to the final release profiles of payloads from these stimuli-responsive nanocapsules. MBT exhibited a burst release over 45% from silica capsules due to the diffusion from the porous shell. The burst release of MBT from silica capsules can be suppressed by using a less porous polymer as the shell material. Remarkably, silica capsules containing tetrasulfide linkage exhibited 100% release upon reduction compared with 70% in the case of silica capsules with disulfide linkage. Only ∼10% of the encapsulated anti-inflammatory drug IMC was leaking from silica nanocapsules after 30 h due to the higher molecular weight of IMC compared to MBT. In fact, the size of IMC was large enough to hinder its diffusion through the reduced tetrasulfide linkages, therefore no increase of IMC release was induced by the cleavage of disulfide or tetrasulfide linkages.The release profiles indicated that (i) the porosity of the shell determined the leakage of payloads, (ii) the cleavage of the redox-responsive linkages in the shell induced the stimuli-specific release of payloads, and (iii) the dimensions of these redox-responsive linkages influenced strongly on the release of payloads with different molecular weights. The investigation of influencing factors of the encapsulation and release of payloads provides fundamental information for the design of stimuli-responsive nanocontainers with switchable/cleavable gates in the shell. In terms of the change of electrochemical potential during the corrosion process of metallic materials and the potential difference between the oxidizing extracellular and reducing intracellular environments,52 the redox-responsive silica and polymer nanocapsules are promising candidates for self-healing in corrosion protection and drug delivery.
Acknowledgements
S. Jiang acknowledge the financial support from “MPG-CAS Joint Doctoral Promotion Program (DPP)”.References
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- D. A. Lavan, T. McGuire and R. Langer, Nat. Biotechnol., 2003, 21, 1184–1191 CrossRef CAS PubMed.
- T. H. Tran, A. Vimalanandan, G. Genchev, J. Fickert, K. Landfester, D. Crespy and M. Rohwerder, Adv. Mater., 2015, 27, 3825–3830 CrossRef CAS PubMed.
- M. Zheludkevich, J. Tedim and M. Ferreira, Electrochim. Acta, 2012, 82, 314–323 CrossRef CAS.
- D. Shchukin and H. Möhwald, Science, 2013, 341, 1458–1459 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Y. Zhao, L.-P. Lv, S. Jiang, K. Landfester and D. Crespy, Polym. Chem., 2015, 6, 4197–4205 RSC.
- C. Huang, S. J. Soenen, J. Rejman, B. Lucas, K. Braeckmans, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2011, 40, 2417–2434 RSC.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- R. H. Staff, D. Schaeffel, A. Turshatov, D. Donadio, H. J. Butt, K. Landfester, K. Koynov and D. Crespy, Small, 2013, 9, 3514–3522 CrossRef CAS PubMed.
- J. Fickert, M. Makowski, M. Kappl, K. Landfester and D. Crespy, Macromolecules, 2012, 45, 6324–6332 CrossRef CAS.
- L.-P. Lv, Y. Zhao, N. Vilbrandt, M. Gallei, A. Vimalanandan, M. Rohwerder, K. Landfester and D. Crespy, J. Am. Chem. Soc., 2013, 135, 14198–14205 CrossRef CAS PubMed.
- R. H. Staff, M. Gallei, K. Landfester and D. Crespy, Macromolecules, 2014, 47, 4876–4883 CrossRef CAS.
- H. C. Chiu, Y. W. Lin, Y. F. Huang, C. K. Chuang and C. S. Chern, Angew. Chem., Int. Ed., 2008, 47, 1875–1878 CrossRef CAS PubMed.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song and A. G. Schwartz, Nat. Mater., 2009, 8, 935–939 CrossRef CAS PubMed.
- L.-P. Lv, K. Landfester and D. Crespy, Chem. Mater., 2014, 26, 3351–3353 CrossRef CAS.
- Y. Zhao, R. Berger, K. Landfester and D. Crespy, Small, 2015, 11, 2995–2999 CrossRef CAS PubMed.
- Y. Ma, W.-F. Dong, M. A. Hempenius, H. Möhwald and G. J. Vancso, Nat. Mater., 2006, 5, 724–729 CrossRef CAS PubMed.
- M. Gallei, Macromol. Chem. Phys., 2014, 215, 699–704 CrossRef CAS.
- R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. D. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS PubMed.
- X. Sui, M. A. Hempenius and G. J. Vancso, J. Am. Chem. Soc., 2012, 134, 4023–4025 CrossRef CAS PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- W. Ong, Y. Yang, A. C. Cruciano and R. L. McCarley, J. Am. Chem. Soc., 2008, 130, 14739–14744 CrossRef CAS PubMed.
- C. Wang, Y. Guo, Y. Wang, H. Xu and X. Zhang, Chem. Commun., 2009, 5380–5382 RSC.
- A. Napoli, M. Valentini, N. Tirelli, M. Müller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- J. Liu, Y. Pang, Z. Zhu, D. Wang, C. Li, W. Huang, X. Zhu and D. Yan, Biomacromolecules, 2013, 14, 1627–1636 CrossRef CAS PubMed.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS PubMed.
- J. Xuan, D. Han, H. Xia and Y. Zhao, Langmuir, 2013, 30, 410–417 CrossRef PubMed.
- W. Chen, P. Zhong, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef CAS PubMed.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS PubMed.
- Y. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728 CrossRef CAS.
- A. N. Koo, H. J. Lee, S. E. Kim, J. H. Chang, C. Park, C. Kim, J. H. Park and S. C. Lee, Chem. Commun., 2008, 6570–6572 RSC.
- J. Fickert, D. Schaeffel, K. Koynov, K. Landfester and D. Crespy, Colloid Polym. Sci., 2014, 292, 251–255 CAS.
- M. Zhao, A. Biswas, B. Hu, K.-I. Joo, P. Wang, Z. Gu and Y. Tang, Biomaterials, 2011, 32, 5223–5230 CrossRef CAS PubMed.
- K. Liang, G. K. Such, Z. Zhu, Y. Yan, H. Lomas and F. Caruso, Adv. Mater., 2011, 23, H273–H277 CrossRef CAS PubMed.
- Y. Yan, Y. Wang, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2011, 23, 3916–3921 CrossRef CAS PubMed.
- G. L. Li, Z. Zheng, H. Möwald and D. G. Shchukin, ACS Nano, 2013, 7, 2470–2478 CrossRef CAS PubMed.
- M. Ejaz, H. Yu, Y. Yan, D. A. Blake, R. S. Ayyala and S. M. Grayson, Polymer, 2011, 52, 5262–5270 CrossRef CAS.
- Y.-J. Pan, Y.-Y. Chen, D.-R. Wang, C. Wei, J. Guo, D.-R. Lu, C.-C. Chu and C.-C. Wang, Biomaterials, 2012, 33, 6570–6579 CrossRef CAS PubMed.
- M. Ohsawa and W. Suëtaka, Corros. Sci., 1979, 19, 709–722 CrossRef CAS.
- W. Konigsberg, Methods Enzymol., 1972, 25, 185–188 CAS.
- S. Budavari, M. J. O'Neil, A. Smith and P. E. Heckelman, The merck index, Merck Rahway, NJ, 1989, vol. 11 Search PubMed.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- S. Jiang, L. Lv, Q. Li, J. Wang, K. Landfester and D. Crespy, Nanoscale, 2016, 8, 11511–11517 RSC.
- R. Woods, G. A. Hope and K. Watling, J. Appl. Electrochem., 2000, 30, 1209–1222 CrossRef CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22733c |
| This journal is © The Royal Society of Chemistry 2016 |