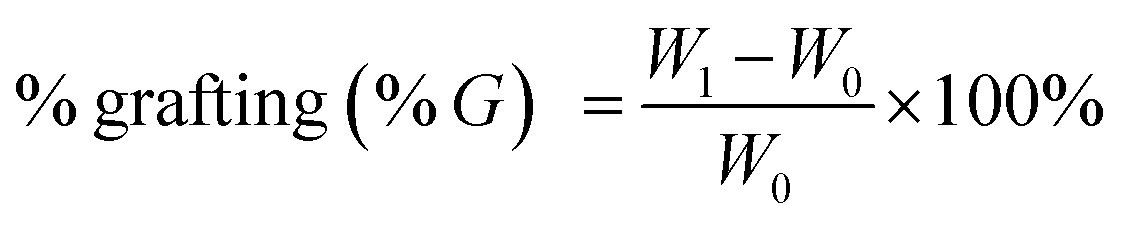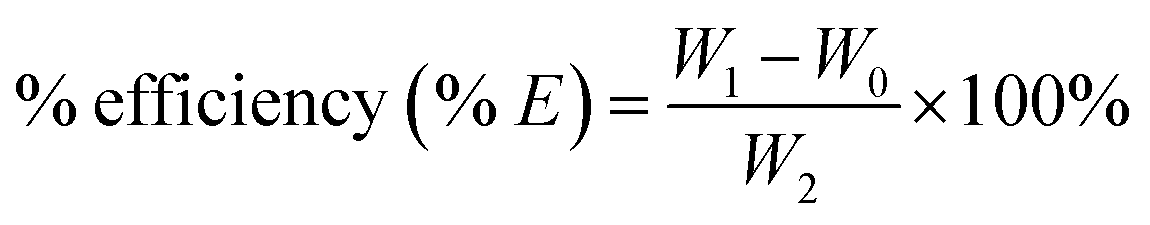DOI:
10.1039/C6RA22730A
(Paper)
RSC Adv., 2016,
6, 110722-110732
Preparation and characterization of temperature-memory nanoparticles of MIP-CS-g-PMMA
Received
12th September 2016
, Accepted 30th October 2016
First published on 8th November 2016
Abstract
A molecular imprinted graft copolymer of chitosan with methyl methacrylate (MIP-CS-g-PMMA) was prepared by free radical polymerization with aspirin as the template molecule and ammonium persulfate as the initiator. The molecular imprinted polymers (MIPs) were characterized by FTIR, X-ray diffraction, thermo-gravimetric analysis, 1H NMR, TEM and SEM. The mechanism of graft copolymerization and factors affecting the graft reaction were studied in detail, and the optimum reaction conditions (to the highest % G and % E as the standard) were obtained at a methylmethacrylate concentration of 1.2 mol L−1, chitosan concentration of 16.67 mol L−1, initiator concentration of 0.0062 mol L−1, temperature of 60 °C and reaction time of 7 h. MIP-CS-g-PMMA had not only the properties of high efficiency selective recognition adsorption to the template molecules, but also of temperature memory, which was a new type of functional polymer material.
1. Introduction
Molecular imprinting is one of the most promising technologies for the preparation of intelligent polymer materials with specific recognition capacities for template molecules.1–5 In recent years, molecularly imprinted polymers (MIPs) have attracted much attention because of their outstanding advantages, such as predetermined recognition ability, stability, relative ease and low cost of preparation, and potential application to a wide range of target molecules.6–8 MIPs were used as chromatographic stationary phases, for enantiomeric separation and for solid-phase extraction (SPE) and also as receptors, antibody and enzyme mimics.9–14 MIPs are synthetic polymers possessing specific cavities designed for a target molecule. In the most common preparation process, monomers form a complex with a template molecule through covalent or non-covalent interactions and are then joined by using a cross-linking agent. After removing the imprinting molecules by chemical reaction or extraction, binding sites are exposed which are complementary to the template molecule in size, shape, and position of the functional groups, and consequently allow their selective adsorption.
Chitosan, is the only amino polysaccharide distributed in large amounts in nature.15 As a natural polysaccharide, chitosan has a variety of excellent properties, such as good biocompatibility, biodegradability and non-toxicity, which has been widely used in agriculture, textile, pharmaceutical and medical field, environmental protection and wastewater treatment.16–22 However, chitosan is insoluble neither in water nor in common organic solvents, which restricts its application in many fields. Due to the presence of –OH and –NH2 on the backbone, it could be introduced different groups onto the repeating units of chitosan by chemical modification. After modification, the solubility of chitosan can be improved and some desired properties can be introduced, and then the application fields of chitosan may be enlarged.23 Therefore, the chemical modification of chitosan is one of the most active areas of research.
Recently, although some works concerning the combination of chitosan in molecular imprinting have been reported,24–28 a few studies on the preparation of MIPs with chitosan grafted poly(methyl methacrylate) and the adsorption properties of molecular recognition can be found. In addition, there was no study reported about temperature memory. Hence, MIPs should be a potential polymer material for wider applications.
The purpose of this study was to prepare a kind of chemical modification of chitosan by using the method to the backbone of chitosan and adopting molecular imprinting technique of graft polymerization. The molecular imprinted polymer of chitosan-g-poly(methyl methacrylate) (MIP-CS-g-PMMA) were analyzed by FTIR, XRD, TGA and 1H NMR. The morphology of the polymer was observed by TEM and SEM. The reaction conditions at the highest % G and % E was optimized and the mechanism of graft reaction was also investigated. With aspirin (ASP) as the template molecule, chloramphenicol and tetracycline for comparison, the molecular imprinted features were explored, also. The properties of molecular recognition and temperature memory of adsorption were studied in detail.
2. Experimental
2.1. Materials
Chitosan (CS, deacetylation degree of 90%) was purchased from Yuhuan Factory, China. Ammonium persulfate and methyl methacrylate (MMA) of analytical grade with purity of 98% were purchased from Company of Tianjin Chemical Reagent. Aspirin (ASP), chloramphenicol and tetracycline with the purity of 95% were provided by Beijing Soledad Bao Technology Co. Ltd., China. All other chemicals were of analytical grade. All commercially solvents and reagents were used without further purification.
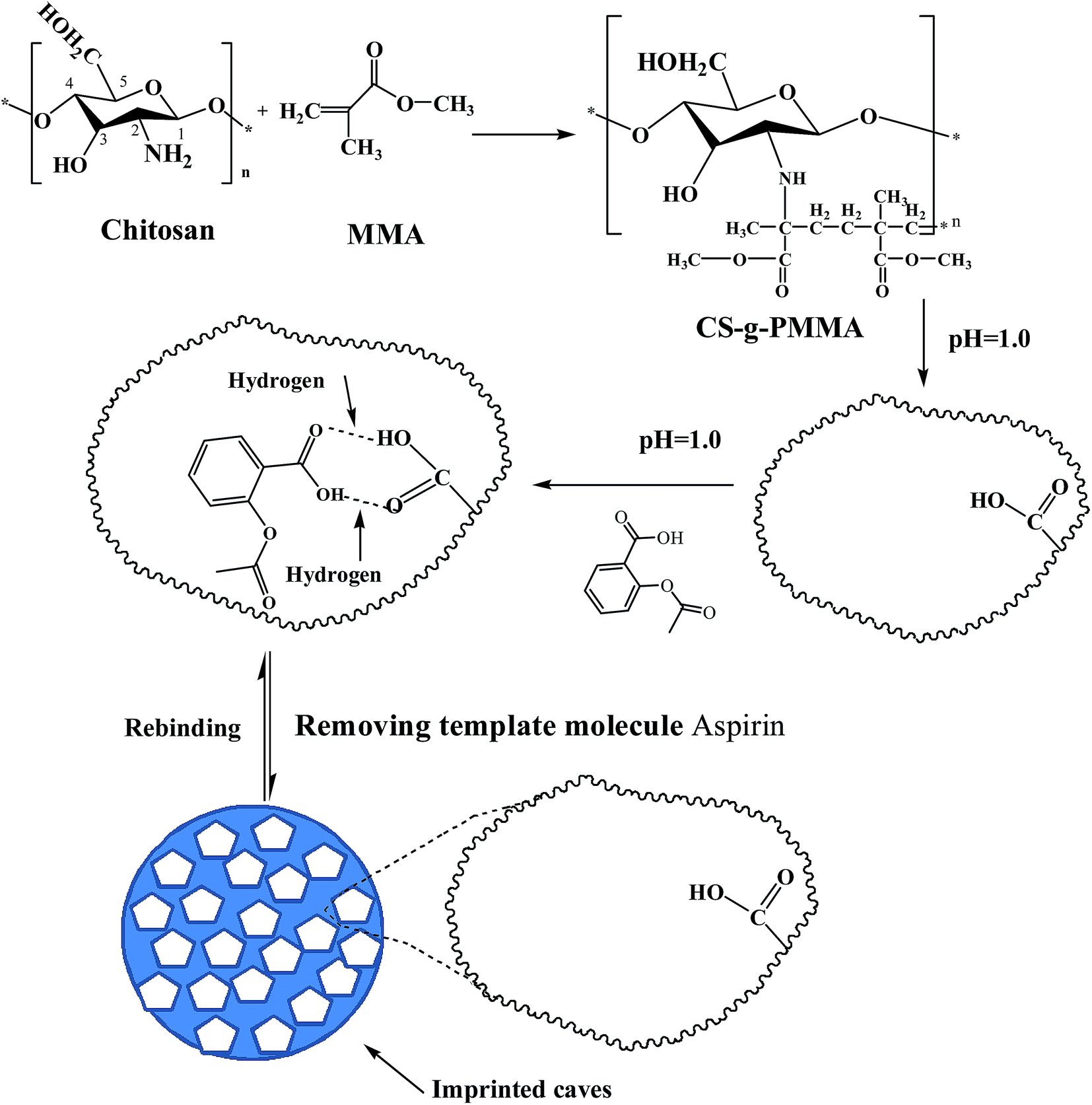
2.2. Synthesis of molecular imprinting polymer chitosan-g-poly(methylmethacrylate) (MIP-CS-g-PMMA)
The general procedure preparing MIP-CS-g-PMMA was depicted as Scheme 1. A desired quantity of chitosan and 3 wt% acetic acid aqueous solution (with the total volume of the aqueous solution 60 mL) were filled in a 250 mL three necked flask. After chitosan was dissolved in the acetic acid aqueous solution, the temperature of the system was controlled at a desired temperature. Ammonium persulfate (0.1 wt%, 8.5 mL) was added into the above system and the mixture was stirred for 30 min and a desired volume of MMA was added to the graft copolymerization reaction. After 30 min, acid solution (pH = 1.0) was added to the above system. After 5 min, a desired quantity of aspirin (used for template molecule) was added into the solution under acidic conditions (pH = 1).
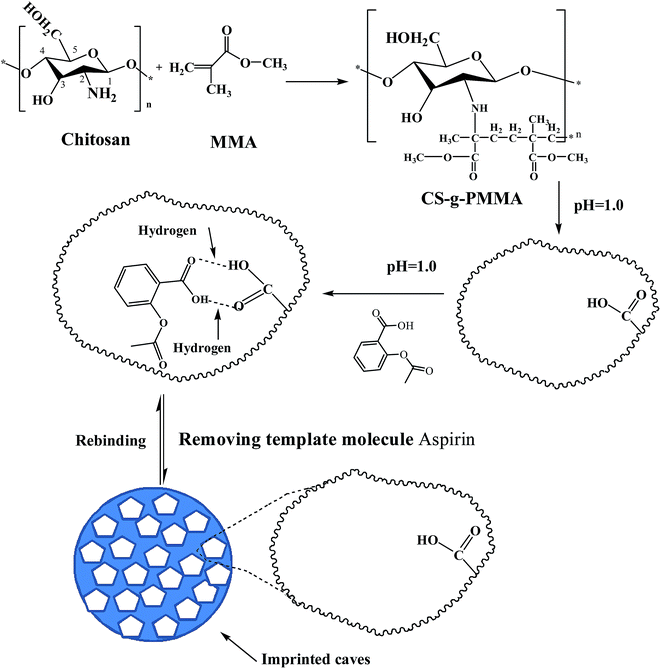 |
| | Scheme 1 Schematic expression of chemical reaction process to prepare imprinted microspheres MIP-CS-g-PMMA. | |
At the time of generating white nanoparticles, the system was continuously stirred for a desired time, and then the mixture was centrifuged to obtain solid. The solid product was washed by distilled water and acetone repeatedly until pH value reached 7, then washed by anhydrous alcohol to remove the salts and small molecular substances. After centrifuging for 3 times, the production was dried at 60 °C to a constant weight. Finally, MIP-CS-g-PMMA was obtained.
For comparison, non-molecularly imprinted polymers (NIP-CS-g-PMMA) was prepared in the absence of template molecule (aspirin) during the polymerization process and treated in the identical manner.
All the samples were dried before used for characterization. Grafting rate (% G), which represents the backbone of the graft polymer in the amount of substrate (chitosan), and efficiency of grafting (% E), which indicates the initial conversion of MMA to the grafted PMMA, were calculated from the increase in weight of chitosan after graft copolymerization using the following formula:29
| |
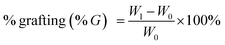 | (1) |
| |
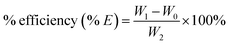 | (2) |
where
W1,
W0 and
W2 denote the weight of the grafted chitosan, the weight of original chitosan and the weight of the used monomer, respectively.
2.3. Characterization
2.3.1 Fourier transform infrared spectroscopy (FT-IR). All FT-IR spectroscopic measurements were performed on a Bruker IFS 66/S spectrometer (Germany). The sample powders were dried and mixed with KBr and pressed into a plate for measurement.
2.3.2 Scanning electron microscopy (SEM) analysis. The surface of the CS-g-PMMA particles is detected by the scanning electron microscope (SEM, Kyky-2800, Kyky technology Co., Ltd).
2.3.3 Transmission electron microscope (TEM) analysis. The size and morphology of CS-g-PMMA nanoparticles were characterized by transmission electron microscope (TEM, H-7650, Hitachi Inc.). The sample of CS-g-PMMA nanoparticles for TEM analysis was obtained by placing a drop of the nanoparticle dispersed ethyl solution onto a copper micro-grid and evaporated in 20 °C.
2.3.4 Thermo-gravimetric analysis (TGA). TGA was carried out on Perkin-Elmer Pyria TGA in N2 atmosphere temperature range of 40–800 °C, heating rate of 10 °C min−1.
2.3.5 Proton nuclear magnetic resonance (1H-NMR). High-resolution 1H-NMR spectra were recorded on a Varian Unity INOVA-400 spectrometer. Samples were prepared as described by Y. Wan et al.32 Native or modified chitosan were dissolved in deuterated water (D2O) with CH3COOD. These solutions were then freeze/thawed three times to exchange labile proton with deuterium and their spectra were recorded at 330 K.
2.3.6 X-ray powder diffraction (XRD). The structure of the membranes samples was characterized by XRD (Philips, Holland). The X-ray diffraction patterns were taken from 10° to 60° (2θ value) using Cu Kα radiation with an intensity ratio (α1/α2) = 0.5 and wavelengths of 1.54439 and 1.54056 Å, respectively.
2.4. Selective adsorption experiments
The adsorption capacity is an important factor to evaluate the special binding and selective recognition of MIPs. Samples (MIPs and NIPs) with the weight of 50 mg were immersed in 500 mL distilled water containing 0.1 g of ASP, chloramphenicol, and tetracycline, respectively. The equilibrium adsorption capacity of MIPs and NIPs to different substrate was determined and defined as Q (mg g−1), which was calculated by formula (3). For comparison, the adsorption capacity of NIP-CS-g-PMMA was also investigated with the same method described above.where C0 (mg L−1) is the initial concentration of the solution; Cs (mg L−1) is the substrate concentration of the solution after adsorption equilibrium; V (mL) is the volume of the initial solution; m (g) is the weight of the polymers.
3. Results and discussions
3.1. Characterization of the MIP-CS-g-PMMA
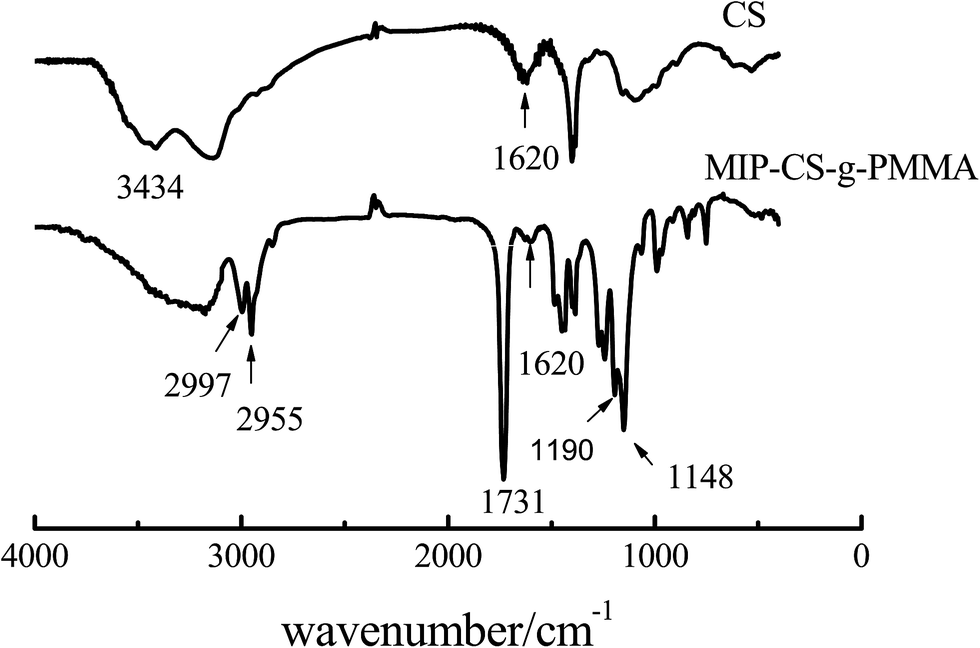
3.1.1 FT IR analysis. The polymer of MIP-CS-g-PMMA was prepared by using chitosan and methyl methacrylate with aspirin as the template molecule. Fig. 1 showed the IR spectra of chitosan and MIP-CS-g-PMMA. In the spectrum of chitosan (CS), the strong peak at 3434 cm−1 was attributed to the stretching vibration of O–H. In the spectrum of MIPs, the sharp absorption peaks of MIP-CS-g-PMMA at 2955 and 2997 cm−1 were assigned to the carbonyl stretching and symmetrical and asymmetrical stretching of the methyl group, respectively.30 The peaks at 1620 cm−1 (due to N–H plane deformation vibration absorption) became weak, the peaks at 1148 cm−1 (assigned to C–O–C stretching) and the peaks at 1731 cm−1 (assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) became significantly enhanced. Furthermore, the peak at 1190 cm−1 was attributed to the C–N stretching vibration. These provided evidences that the PMMA had grafted onto the chitosan. As for the bands in the region of 3000–3500 cm−1, which was became broader in the spectrum of copolymerization than chitosan, indicating that there were more hydrogen bonds in the MIPs and this could generate advanced performance of molecular imprinting.
O stretching) became significantly enhanced. Furthermore, the peak at 1190 cm−1 was attributed to the C–N stretching vibration. These provided evidences that the PMMA had grafted onto the chitosan. As for the bands in the region of 3000–3500 cm−1, which was became broader in the spectrum of copolymerization than chitosan, indicating that there were more hydrogen bonds in the MIPs and this could generate advanced performance of molecular imprinting.
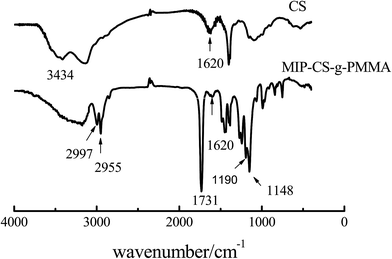 |
| | Fig. 1 FTIR spectra of chitosan and MIP-CS-g-PMMA. | |
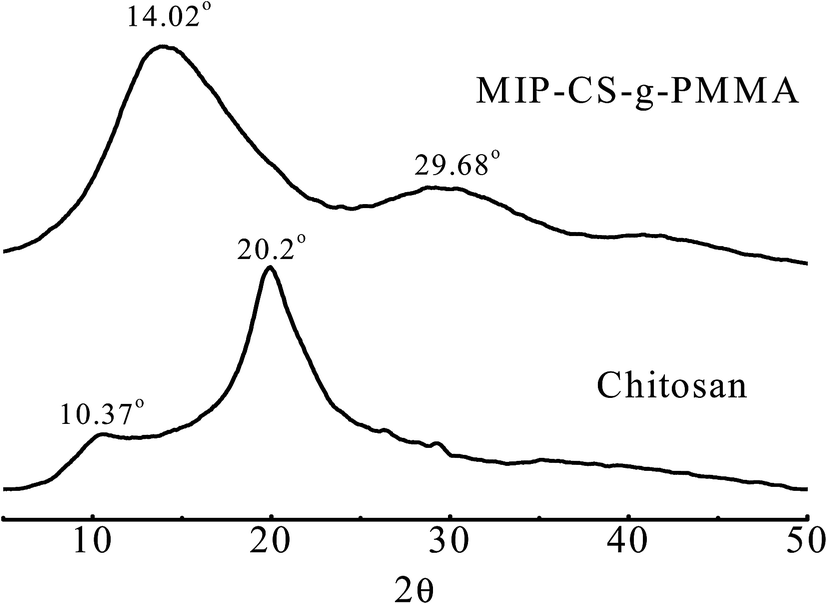
3.1.2 XRD analysis. The structure of the membrane samples was characterized by XRD which were taken from 10° to 60° (2θ value). Fig. 2 showed the X-ray diffraction (XRD) pattern of MIPs and chitosan.31 The crystalline peaks (2θ) at 10.3° and 20.2° were attributed to chitosan, which are typical fingerprint for chitosan and very similar to the work of Wan, Creber, Preppley, and Bui.32 From previous report,33 the small peak at approximately 10° (2θ) was attributed to the anhydrous crystal of chitosan whereas the diffraction peak at around 21–22° (2θ) was observed in chitosan prepared from dissolving chitosan in acetic acid solution34 While after PMMA grafting, it acquires crystalline areas around 14.02° (2θ) and 30° (2θ) (Fig. 2) in the copolymerization. This was because numerous of hydrogen bonds formed to destruct the original crystalline structure of the chitosan during the synthesis, which indicated that the PMMA had grafted onto chitosan successfully.
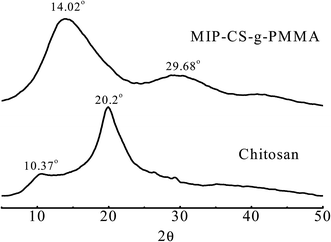 |
| | Fig. 2 X-Ray diffractograms of chitosan and MIP-CS-g-PMMA. | |
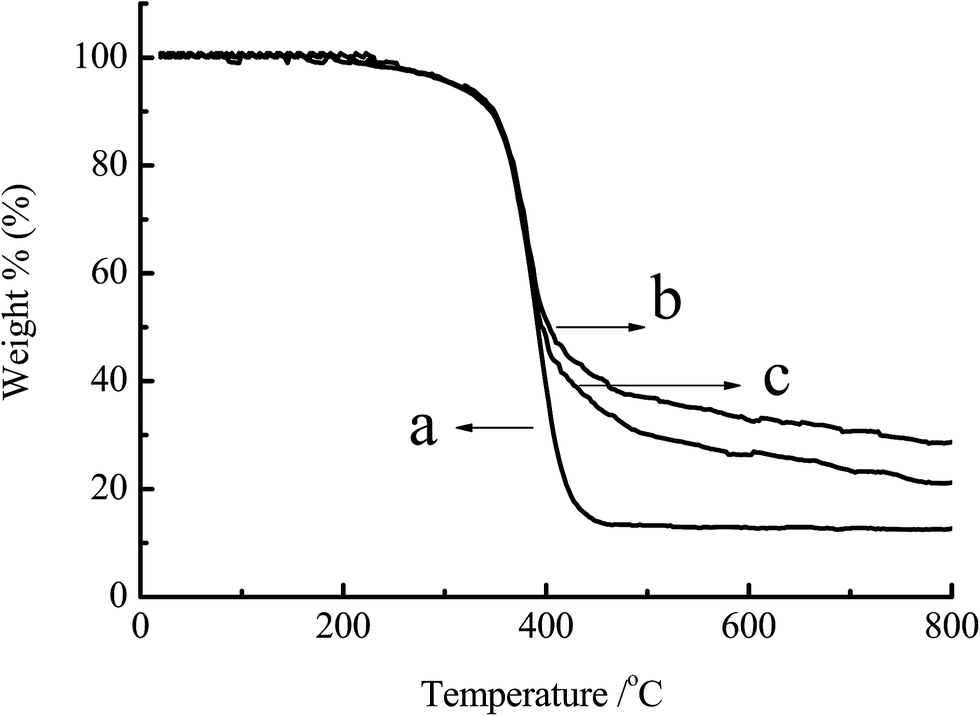
3.1.3 TGA analysis. The power samples of chitosan, CS-g-PMMA (% E = 47) and CS-g-PMMA (% E = 21) were dried in the vacuum oven and analyzed by TGA. Fig. 3 showed the TGA curves of the chitosan and CS-g-PMMA with different efficiency of grafting. Curves of DTG derived from the derivation of TGA indicated that thermal decomposition temperature of curve (a) was around 430 °C, while curve (b) and (c) was approximately 410 °C. Thermal decomposability had a little difference between (a), (b) and (c). However, as shown in Fig. 3, the residual of the three samples was significantly different: (a) 10%, (b) 27% and (c) 21%, respectively. The reason may be that the process of TGA in N2 atmosphere was accompanied with the carbonization of PMMA. In the range of 0–410 °C, thermal decomposition occurred in all the three samples, but within the range of 410–800 °C, carbonization did not occur in (a) but happened in the graft copolymer (b) and (c) due to the presence of PMMA. The more the % E, the higher the residual rate.35 This further confirmed the success of grafting reaction.
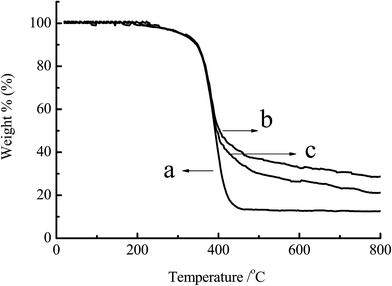 |
| | Fig. 3 TGA for (a) original chitosan, (b) CS-g-PMMA (% E = 47) and (c) CS-g-PMMA (% E = 21). | |
3.1.4 1H NMR analysis. Original and polymer of modified chitosan were dissolved at a concentration of 2% in deuterated water (D2O) with CH3COOD. These solutions were then freezed/thawed three times to exchange labile proton with deuterium and their spectra were recorded at 330 K. Fig. 4 displayed the 1H NMR spectra of (a) original chitosan, (b) pure PMMA and (c) copolymerization CS-g-PMMA with % E = 47. In Fig. 4(a), the peaks at 0.67 and 1.4 ppm were attributed to the CH3 of chitosan, and the peaks at 2.1 ppm was ascribed to the H–N of chitosan. The H-3 and H-4 protons are found around 3.7 ppm and 3.5 ppm, respectively. In spectrum (b), the peaks at 1.86, 2.02 and 3.64 ppm were assigned to the –CH3 of PMMA, and the peaks at 5.85 and 6.20 ppm to –CH2–. By comparison, new peaks at 0.7, 0.9, and 1.9 ppm in Fig. 4(c) were ascribed to CH3, –CH2–, and –CH2 (COOH) of the copolymerization CS-g-PMMA, respectively. These results also confirmed that the PMMA was grafted to chitosan successfully.
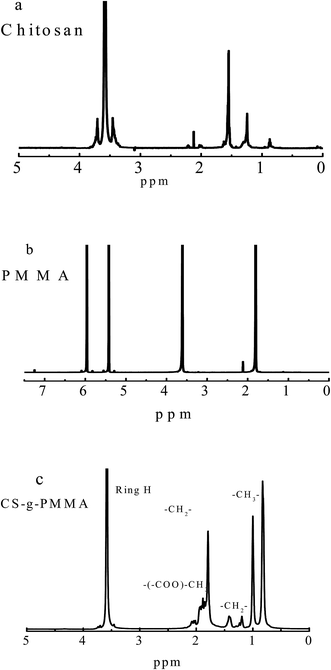 |
| | Fig. 4 1H NMR spectra of (a) original chitosan, (b) pure PMMA and (c) copolymerization CS-g-PMMA (% E = 47) dissolved in D2O. | |
3.1.5 Graft copolymerization morphology analysis. Fig. 5 demonstrated the TEM image (a) and SEM image (b) of MIP-CS-g-PMMA. As shown from image (a) and (b), the graft copolymerization was a unimodal size distribution within 150–200 nm, indicating that the nanoparticles were a relatively homogeneous dispersion.
 |
| | Fig. 5 The TEM image (a) and SEM image (b) of MIP-CS-g-PMMA (template molecule of aspirin had been washed off). | |
3.2. Mechanism of the graft copolymerization
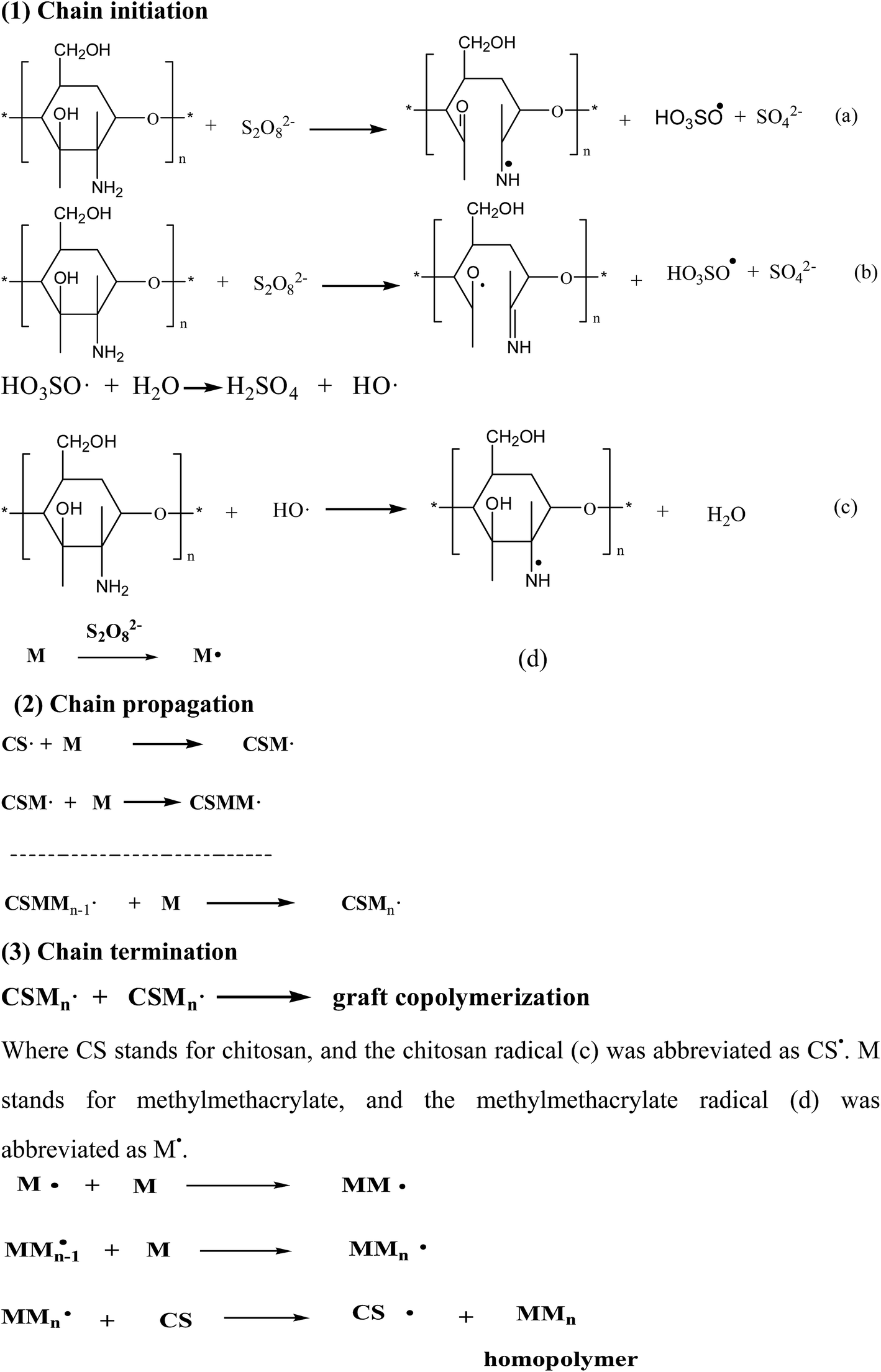
The graft copolymerization of chitosan was formed by three elementary reactions: chain initiation, chain propagation and chain termination, as shown in Scheme 2. Initiator was due to the ammonium persulfate (as an initiator) decomposed which could be achieved by two ways: (1) primary radicals generated by the decomposition made C2 and C3 carbon atoms of chitosan so activated that the bond was broken between C2 and C3, and generated two radicals (a) and (b) in Scheme 2. (2) Free radicals HO3SO˙ generated by ammonium combined with HO˙ formed by water firstly, and then combined with active hydrogen on the amino group of chitosan to form new radical (c). Methylmethacrylate (MMA) could easily form free radicals M˙ (d) in the condition of persulfate ions.36 In this experiment, the main free radicals were (c) and (d) which would be investigated in the further work.
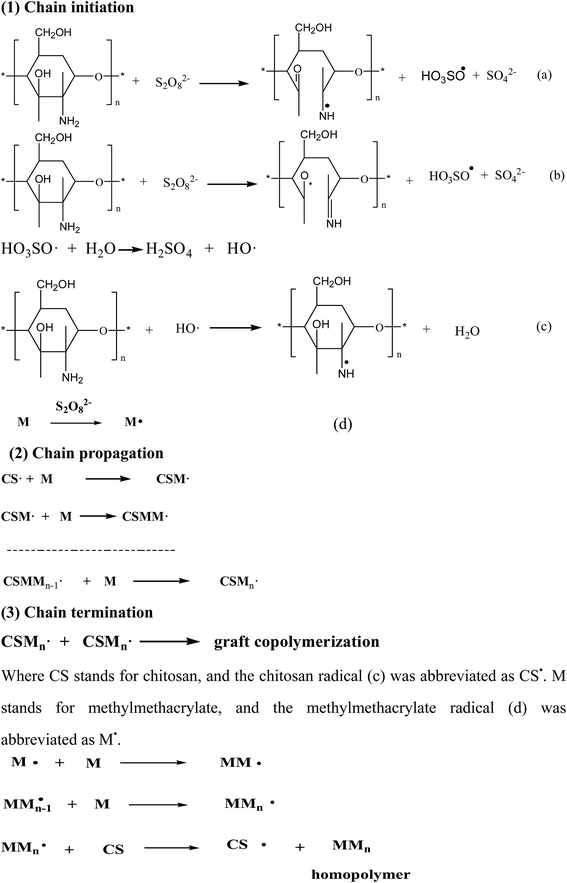 |
| | Scheme 2 Mechanism of the graft copolymerization of chitosan, where CS represents chitosan and M represents MMA monomer. | |
As shown in Scheme 1, the free radicals captured the atom H of –NH2 on the backbone of chitosan37 to form the macromolecular radicals of chitosan, which lead to the graft copolymerization and homopolymerization reactions. And finally grafting reaction was terminated with two macromolecular chain radicals reacting with each other. At the same time, homopolymer of PMMA was generated, indicating the important features of graft copolymerization. And M˙ could collide with MMA to form the free radical of MMn which would collide with CS to generate the free radical of CS and homopolymer of MMn.36
3.3. Effect of the reaction parameters on the graft copolymerization
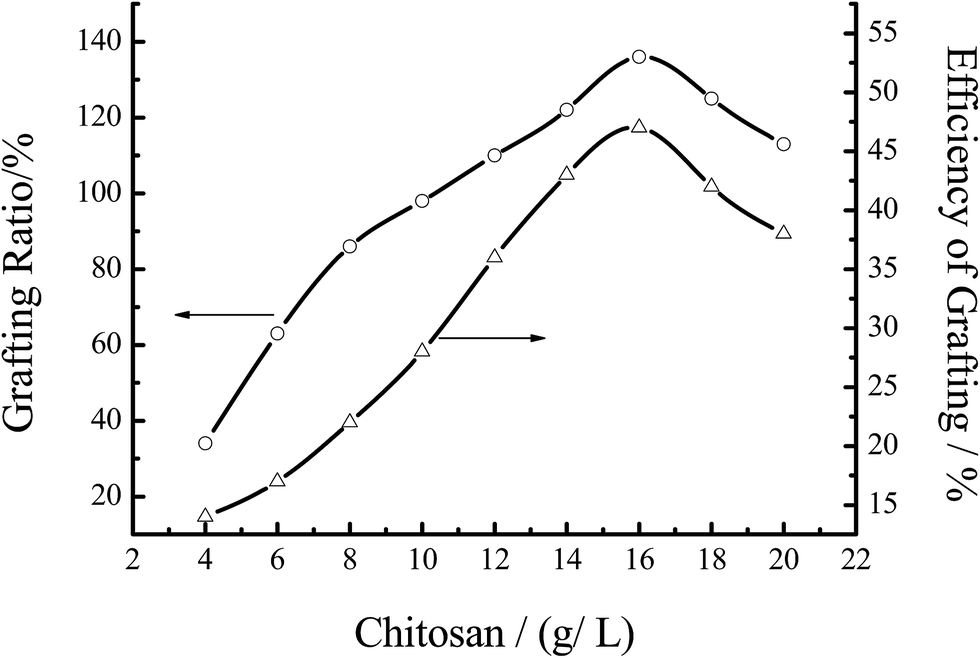
3.3.1 Effect of the chitosan concentration. Fig. 6 showed the effect of the chitosan concentration on the grafting efficiency under the indicated reaction conditions. When the concentration of chitosan increased, the % G and % E both increased immediately. This phenomenon can be explained by the graft mechanism. With increasing the concentration of chitosan, the total number of group –NH2 increased, resulting in more activated sites for MMA to attack the radicals of chitosan. Therefore, % G and % E of the graft copolymerization both increased with increasing the concentration of chitosan. However, when the concentration of chitosan was too large, the system became so thick that the radicals of chitosan could not move freely, resulting in the decrease of % G and % E. So, in the further experiment, the concentration of chitosan was 16.67 g L−1.
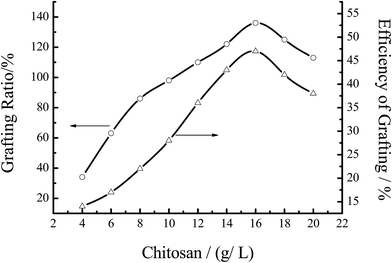 |
| | Fig. 6 Effect of the chitosan concentration on the grafting efficiency. Reaction conditions: [MMA] 1.2 mol L−1, [initiator] 0.0062 mol L−1, temperature 60 °C, reaction time 7 h. | |
3.3.2 Effect of the reaction temperature. The influence of temperature on graft parameters was studied by varying the reaction temperature from 30–80 °C and keeping other parameters fixed. As shown in Fig. 7 at the initial stage of the reaction, % G and % E of the graft copolymerization both increased with increasing the temperature from 30–60 °C, and then decreased. This behavior could be interpreted that the activity of the initiator increased with the increase of temperature, which was helpful to improve the activity of both graft copolymerization and homopolymerization. However, when the temperature exceeded 60 °C, the initiator ability of free radicals reduced with the decrease of the stability of the active center, resulting in decreasing both % G and % E. On the other hand, with increasing the temperature, the mobility of macro radicals would be improved, and more prone to the chain termination at higher temperature.38 Thus, the polymerization activity began to decline, and % G and % E of the graft copolymer were decreased at the elevated temperature.
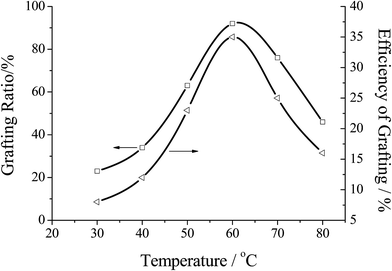 |
| | Fig. 7 Effect of the reaction temperature on the grafting efficiency. Reaction conditions: [MMA] 1.2 mol L−1, [initiator] 0.0062 mol L−1, [chitosan] 16.67 mol L−1, reaction time 7 h. | |
3.3.3 Effect of the reaction time. The influence of reaction time on the graft parameters was investigated by changing the reaction time from 4 to 8 hours, keeping other parameters fixed. As can be seen in Fig. 8, the % G and % E both increased with the increase of reaction time, and then decreased. The maximum grafting ratio was reached when the reaction time was 7 hour. But when the time was more than 7 hour, % G and % E decreased with the increase of the side reactions in the copolymerization.
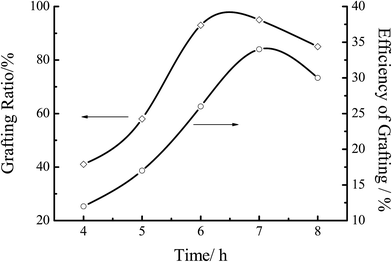 |
| | Fig. 8 Effect of reaction time on the grafting efficiency. Reaction conditions: [MMA] 1.2 mol L−1, [initiator] 0.0062 mol L−1, [chitosan] 16.67 mol L−1, temperature 60 °C. | |
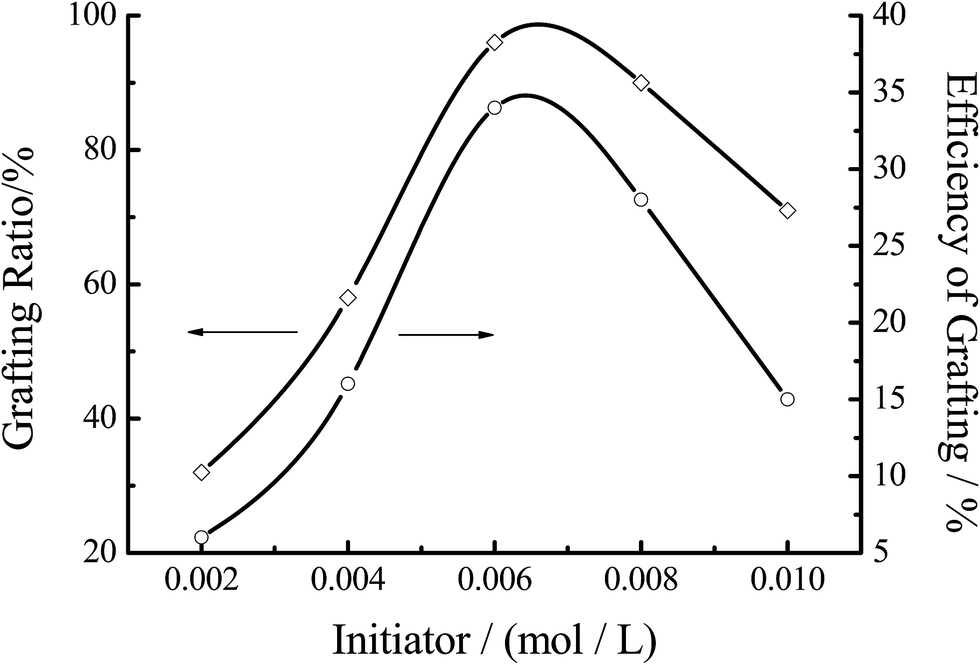
3.3.4 Effect of the initiator concentration. Fig. 9 showed the effect of the initiator concentration on the grafting efficiency. % G and % E both increased with the increase of the initiator concentration at the first stage of the reaction, and then decreased. The maximum of % G was achieved at around 0.0062 mol L−1 of the concentration of the initiator. When the concentration of initiator was 0.002 mol L−1, the grafting efficiency of copolymerization was reduced (about 6.2%). With the initiator concentration increased, the active center formed by the initiator and chitosan increased, resulting in the increase of % G and % E. However, when the concentration was more than 0.006 mol L−1, the excessive free radicals would trigger plurality of coupling, so that the homopolymerization of MMA played a dominant role in the reaction.
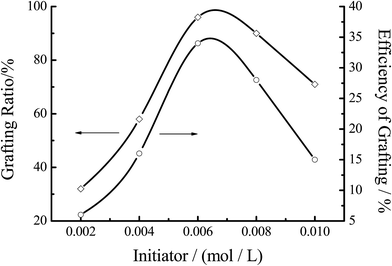 |
| | Fig. 9 Effect of the initiator concentration on the grafting efficiency. Reaction conditions: [MMA] 1.2 mol L−1, [chitosan] 16.67 mol L−1, temperature 60 °C, reaction time 7 h. | |
3.4. Selective adsorption of MIP-CS-g-PMMA
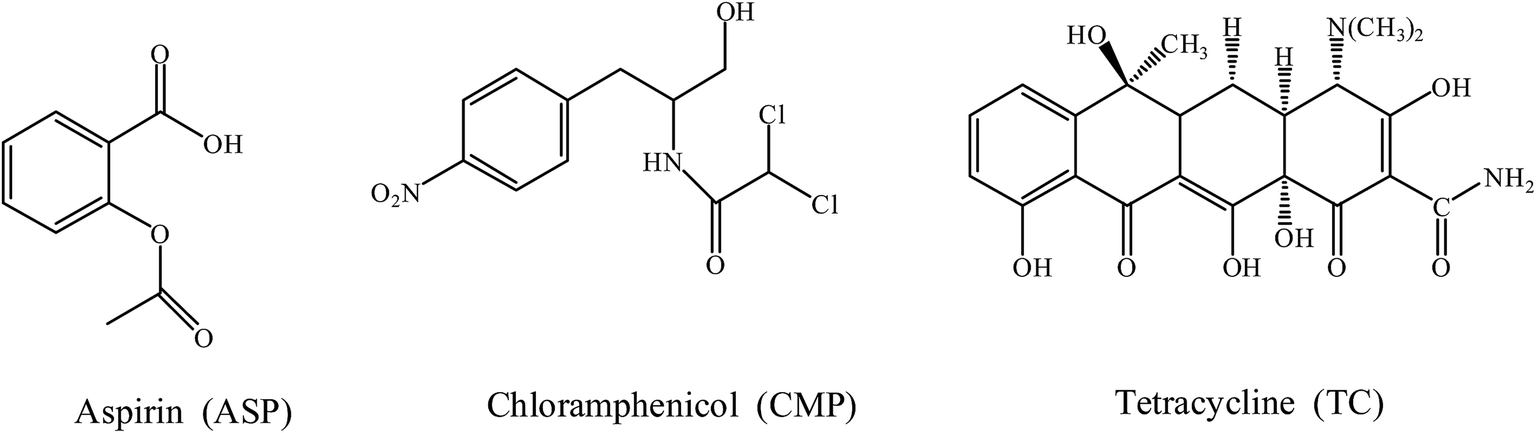
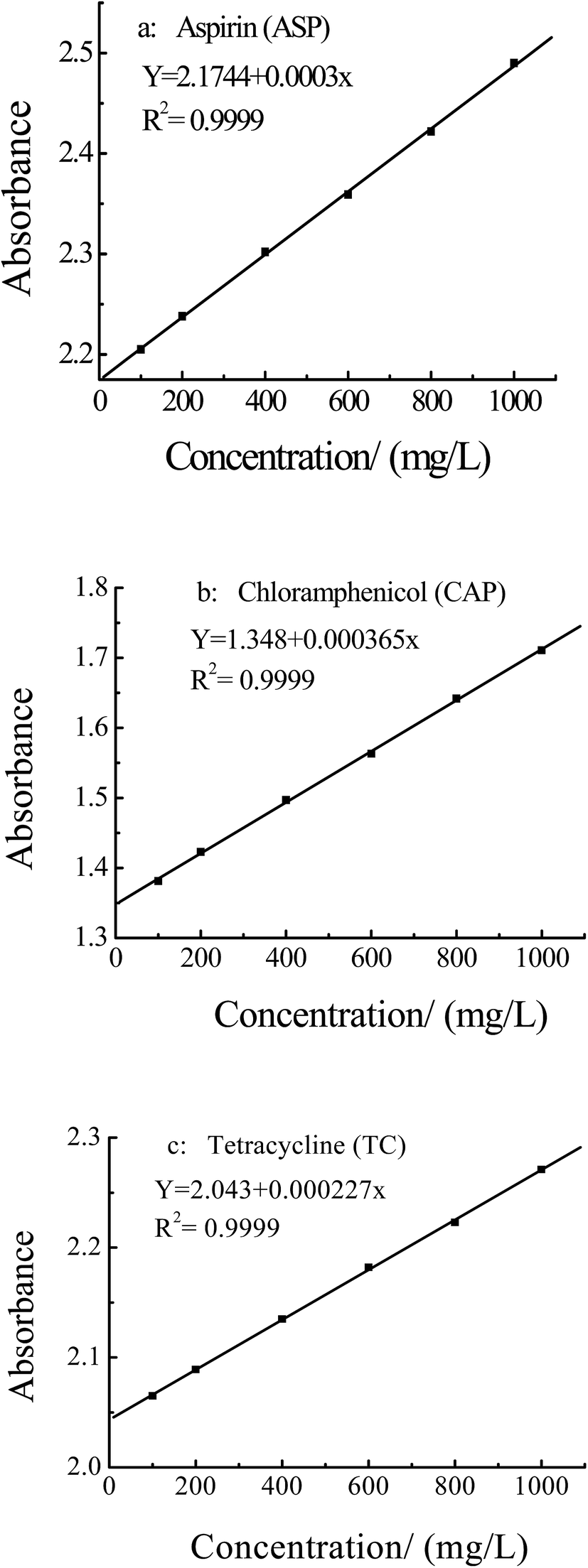
3.4.1 Tests of selective adsorption of different substances. The selectivity of the MIPs was investigated with the ASP template, and CAP and TC as reference compounds, which chemical structures were shown in Fig. 10. The standard curves of ASP, CAP and TC were shown in Fig. 11, and the maximum absorption wavelength (λmax) was 303 nm, 270 nm and 545 nm, respectively.
 |
| | Fig. 10 Chemical structures of template molecular aspirin and the reference compounds (chloramphenicol and tetracycline). | |
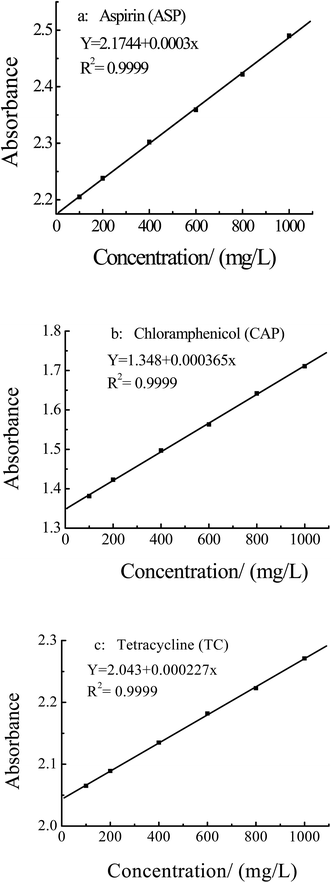 |
| | Fig. 11 Standard curves of aspirin, chloramphenicol and tetracycline solution. | |
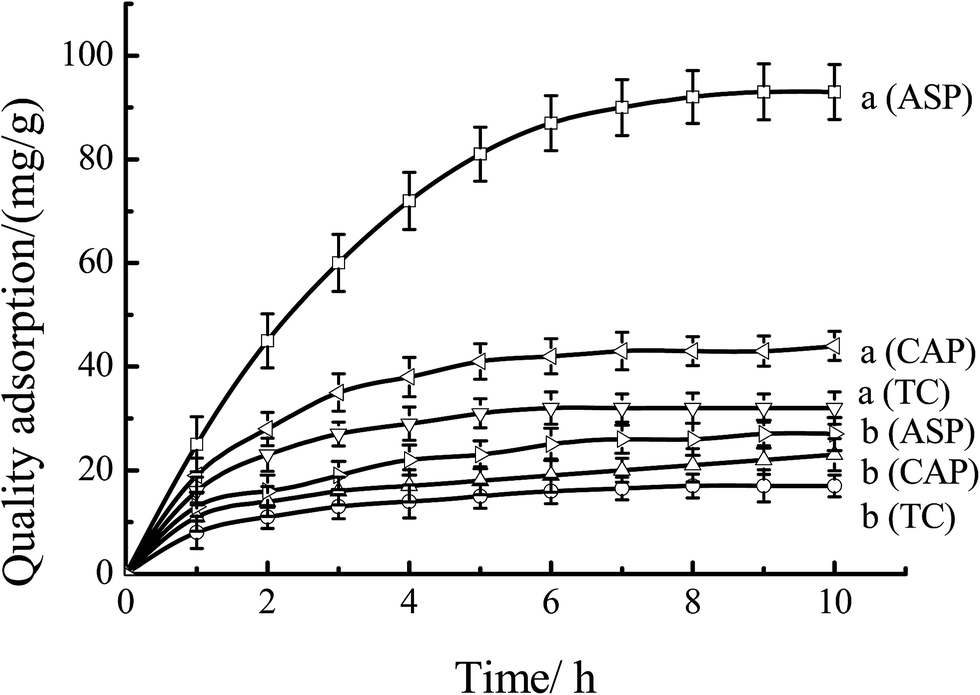
Fig. 12 showed the quality adsorption of the MIPs and NIPs. As shown in Fig. 12, as for MIPs, the quality of adsorption to ASP was much larger than that of CAP and TC. At the time of 8 hours, the quality adsorption to ASP, CAP and TC were 92, 43 and 32 mg g−1, respectively. This behavior can be explained by that the specific recognition capacity of the polymer was achieved by forming a complex between the ASP and the CS-g-PMMA in the MIPs. Removal of ASP from MIPs revealed binding sites that were complementary in size, shape and functional group to the template molecule. As to NIPs, the quality amount of ASP, CAP and TC were only 26, 21 and 17 mg g−1, respectively. This was mainly because the binding sites were not left and the irregular voids were easily blocked by the molecules, resulting in the decrease of the adsorption amount. Therefore, the obtained MIPs have special selectivity to template molecules ASP.
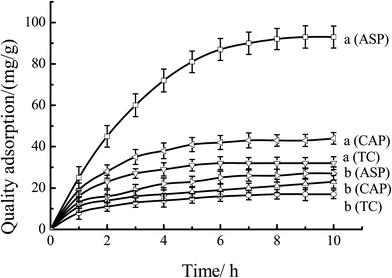 |
| | Fig. 12 Adsorption curves of the mixture solutes through (a) MIP-CS-g-PMMA, and (b) NIP-CS-g-PMMA at 60 °C. | |
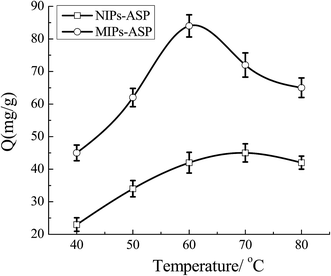
3.4.2 Tests of temperature memory performance of MIP-CS-g-PMMA. As shown in Fig. 13, the effect of temperature on the adsorption amount was studied. For both MIPs and NIPs, the adsorbed amount of ASP increased with the increase of temperature. But the amount of MIPs was much larger that of NIPs, the maximum of 89 mg g−1 was achieved for MIPs at 60 °C. This phenomenon can be explained by that the preparation temperature of the polymer was 60 °C, which was remembered by the cavies in the polymer and the cavies would perfectly match with the template molecule ASP. The complementary shape and the functional group binding sites of the template molecule were generated during the preparation at 60 °C, resulting in the MIPs with high amount adsorption of ASP at the temperature of 60 °C. Therefore, the MIPs remembered the molecular shape at 60 °C, and achieved the molecular imprinting function of temperature sensitivity. As for NIPs, the maximum adsorbed amount of ASP was only 42 mg g−1, which was because that there were no binding sites left which could highly match with template molecular. Thus, the amount of adsorption was low, which was relied on simple physical adsorption.
 |
| | Fig. 13 Effect of temperature on the selective adsorption under different temperature. | |
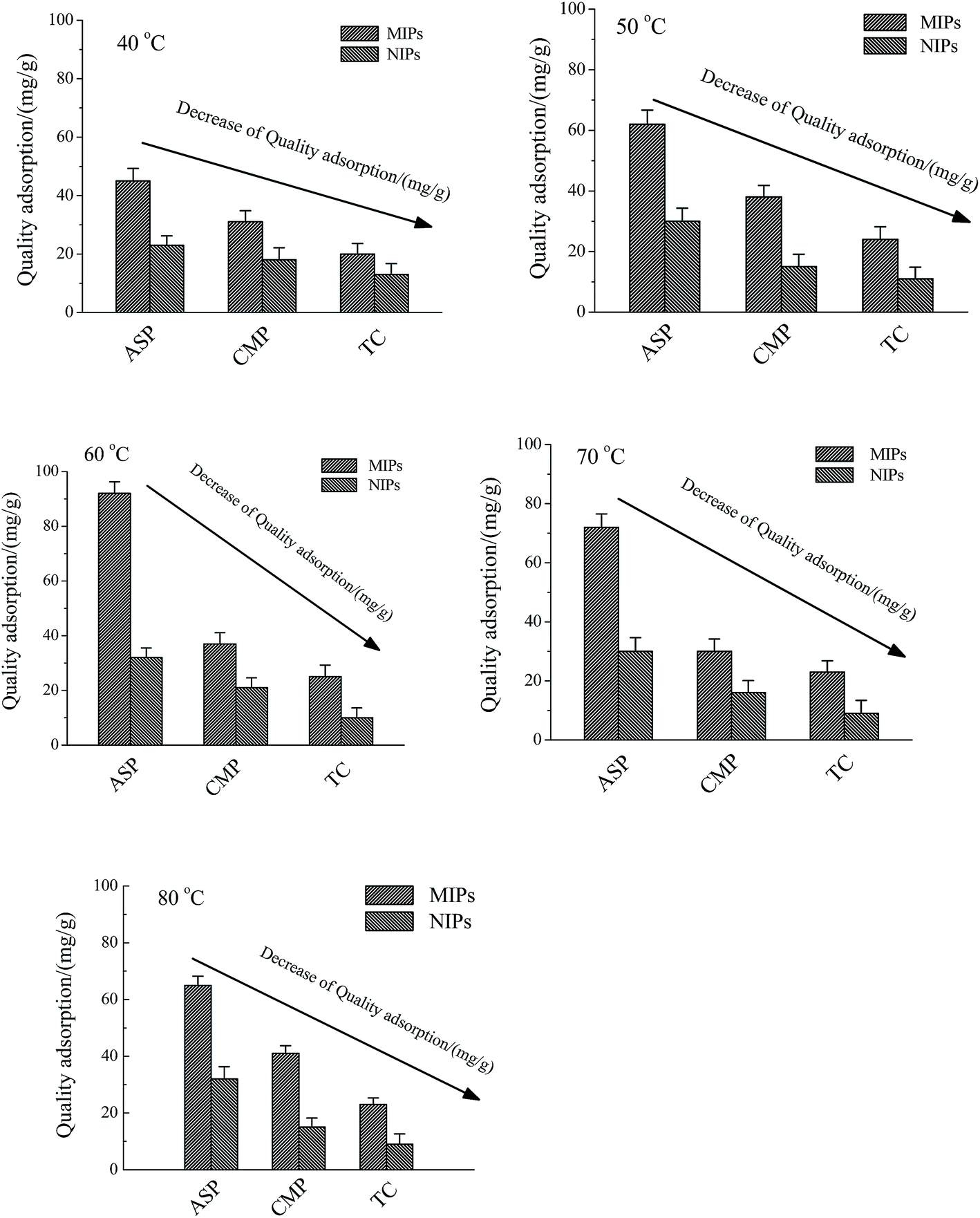
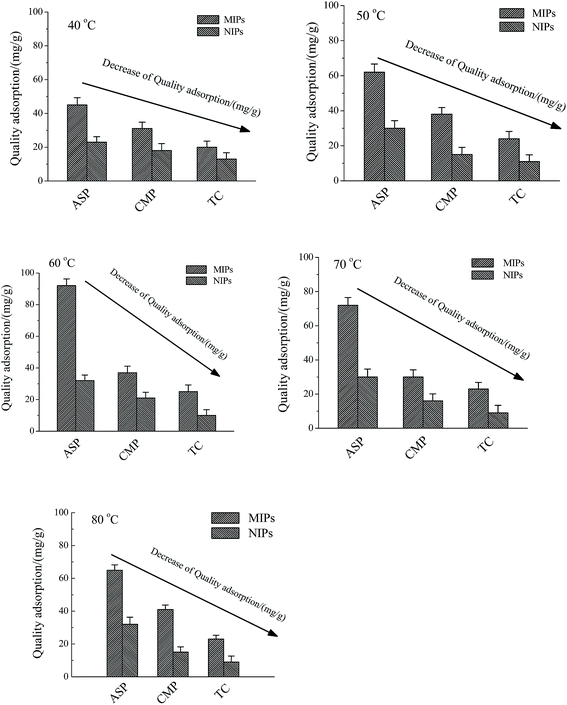
3.4.3 Effect of temperature to the quality adsorption of the ASP, CMP and TC. As for MIP-CS-g-PMMA, the optimum preparation temperature was 60 degrees, and under this temperature the MIP-CS-g-PMMA could exhibit not only the highest E% and G%, but also the lowest energy and the smallest space bit group. This phenomenon can be explained that the template molecules can be firmly bonded in the polymer to form a stable structure in the form of hydrogen bonds at 60 degrees. After removal the template molecules by chemical reaction or extraction, binding sites are exposed which are complementary to the template molecule in size, shape, and position of the functional groups, and consequently allow their high selective adsorption. At different temperature (40, 50, 60, 70, 80 degrees), the adsorption capacity increased from 40 degrees to 60 degrees firstly and then decreased from 60 degrees to 80 degrees. According to the increase of temperature from 40 degrees to 60 degrees, adsorption capacity increased because of the gradually expanding of the imprinted holes, and at 60 degrees (the preparation temperature of MIP-CS-g-PMMA), matching with the template molecules and imprinted holes perfectly, the adsorption capacity to template molecules (ASP) reached the maximum about 92 mg mL−1. With the increase of the temperature from 60 degrees to 80 degrees, the movement of molecules became more and more intense, and the molecules could not be bounded in the imprinted holes, leading to the gradual decline in adsorption capacity. Therefore, the temperature of maximum adsorption capacity was 60 degrees. However, the MIPs adsorption capacity of CMP and TC was smaller than that of SAP, which was due to that the moleculars CMP and TC were bigger than template ASP, leading to not match with the imprinted holes on the surface of the polymer microspheres. As for NIPs, NIPs were synthesized at the same temperature as MIPs, indicating that they have the same trend of adsorption capacity. However, without the imprinted holes, the adsorption capacity of NIPs was smaller than that of MIPs from 40 degrees to 80 degrees (Fig. 14).
 |
| | Fig. 14 Effect of the MIPs and NIPs to the quality adsorption of the ASP, CMP and TC at different temperature (40 °C, 50 °C, 60 °C, 70 °C, and 80 °C). Conditions: methylmethacrylate concentration of 1.2 mol L−1, chitosan concentration of 16.67 mol L−1, initiator concentration of 0.0062 mol L−1, temperature of 60 °C and reaction time of 7 h. | |
4. Conclusions
Molecular imprinting graft copolymer of chitosan and methylmethacrylate was successfully synthesized by free radical polymerization using ammonium persulfate as initiator. The structure and the morphology of the polymer were also analyzed. The optimum reaction conditions at the highest grafting rate and efficiency of grafting rate were obtained at methylmethacrylate concentration of 1.2 mol L−1, chitosan concentration of 16.67 mol L−1, initiator concentration of 0.0062 mol L−1, temperature of 60 °C and reaction time of 7 h. Molecular imprinting graft copolymer exhibited not only the properties of molecular recognition but also the performance of temperature memory, which was a new type of functional polymer material.
Acknowledgements
The authors are grateful for the financial support from Science and Technology Projects of Hebei Province (No. 15211226, 15213618), Scientific Research Project of Higher Education in Hebei Province (No. ZD20131015).
References
- H. H. Yang, S. Q. Zhang, F. Tan, Z. X. Zhuang and X. R. Wang, J. Am. Chem. Soc., 2005, 127, 1378 CrossRef CAS PubMed.
- C. Xie, Z. Zhang, D. Wang, G. Guan, D. Gao and J. Liu, Anal. Chem., 2006, 78, 8339 CrossRef CAS PubMed.
- X. F. Zheng, Q. Lian and H. Yang, et al., Sci. Rep., 2016, 6, 21409 CrossRef CAS PubMed.
- E. Yilmaz, K. Haupt and K. Mosbach, Angew. Chem., Int. Ed., 2000, 39, 2115 CrossRef CAS.
- J. Matsui, M. Higashi and T. Takeuchi, J. Am. Chem. Soc., 2000, 122, 5218 CrossRef CAS.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch and I. A. Nicholls, J. Mol. Recognit., 2006, 19, 106 CrossRef CAS PubMed.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922 RSC.
- E. Turiel and A. Martin-Esteban, Anal. Chim. Acta, 2010, 668, 87 CrossRef CAS PubMed.
- T. Hishiya, H. Asanuma and M. Komiyama, Polym. J., 2003, 35, 440 CrossRef CAS.
- K. Haupt, Analyst, 2001, 126, 747 RSC.
- G. Theodoridis and P. Manesiotis, J. Chromatogr. A, 2002, 948, 163 CrossRef CAS PubMed.
- K. Haupt, Chem. Commun., 2003, 2, 171 RSC.
- J. Svitel, I. Surugiu and A. Dzgoev, J. Mater. Sci.: Mater. Med., 2001, 12, 1075 CrossRef CAS PubMed.
- I. A. Nicholls, J. Matsui, M. Krook and K. Mosbach, J. Mol. Recognit., 1996, 9, 652 CrossRef CAS PubMed.
- R. A. A. Muzzarelli, Carbohydr. Polym., 1996, 29, 309–316 CrossRef CAS.
- A. Grenha, C. I. Grainger, L. A. Dailey, B. Seijo, G. P. Martin and C. R. López, Eur. J. Pharm. Sci., 2007, 31(2), 73 CrossRef CAS PubMed.
- C. C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58(12–13), 1379–1408 CrossRef CAS PubMed.
- A. Khademhosseini and R. Langer, Biomaterials, 2007, 28(34), 5087–5092 CrossRef CAS PubMed.
- S. Y. Ong, J. Wu, S. M. Moochhala, M. H. Tan and J. Lu, Biomaterials, 2008, 29(32), 4323–4332 CrossRef CAS PubMed.
- A. Ghaffari, K. Navaee, M. Oshoui, K. Bayati and M. Rafiee-Tehrani, Eur. J. Pharm. Biopharm., 2007, 67(1), 175–186 CrossRef CAS PubMed.
- X. Wei, N. Sun, B. Wu, C. Yin and W. Wu, Int. J. Pharm., 2006, 318(1–2), 132–138 CrossRef CAS PubMed.
- A. S. Hoffiman, P. S. Stayton, O. Press, N. Murthy, C. A. Lackey and C. Cheung, Polym. Adv. Technol., 2002, 13(10), 992–999 CrossRef.
- R. Jayakumar, M. Prabaharan, R. L. Resi and J. F. Mano, Carbohydr. Polym., 2005, 62(2), 142–158 CrossRef CAS.
- T. Y. Guo, Y. Q. Xia, J. Wang, M. D. Song and B. H. Zhang, Biomaterials, 2005, 26, 5737 CrossRef CAS PubMed.
- G. Z. Kyzas, N. K. Lazaridis and D. N. Bikiaris, Carbohydr. Polym., 2013, 91, 198 CrossRef CAS PubMed.
- P. A. Nishad, A. Bhaskarapillai, S. Velmurugan and S. V. Narasimhan, Carbohydr. Polym., 2012, 87, 2690 CrossRef.
- M. Monier and A. M. A. El-Sokkary, Int. J. Biol. Macromol., 2010, 47, 207 CrossRef CAS PubMed.
- Y. Tong, H. Guan, S. Wang, J. Xu and C. He, Carbohydr. Polym., 2011, 346, 495 CrossRef CAS PubMed.
- J. D. Boonstra and A. Bekker, Ger Pat., 2 44 012, 1975.
- V. Singh, A. K. Sharma, D. N. Tripathi and R. Sanghi, J. Hazard. Mater., 2009, 161, 955–966 CrossRef CAS PubMed.
- X. F. Zheng, Q. Lian and H. Yang, RSC Adv., 2014, 4, 42478–42485 RSC.
- Y. Wan, K. A. M. Creber, B. Preppley and V. T. Bui, Polymer, 2003, 44, 1057–1065 CrossRef CAS.
- P. C. Srinivasa, M. N. Ramesh, K. R. Kumar and R. N. Tharanathan, J. Food Eng., 2004, 63, 79–85 CrossRef.
- G. C. Ritthidej, T. Phaechamud and T. Koizumi, Int. J. Pharm., 2002, 232, 11–22 CrossRef CAS PubMed.
- P. J. Lv, Y. Z. Bin, Y. Q. Li, R. Chen, X. Wang and B. Y. Zhao, Polymer, 2009, 50, 5675–5680 CrossRef CAS.
- V. Singh, D. N. Tripathi, A. Tiwari and R. Sanghi, Carbohydr. Polym., 2006, 65, 35–41 CrossRef CAS.
- V. Singh, A. Tiwari, D. N. Tripathi and R. Sanghi, Polymer, 2006, 47(1), 254–260 CrossRef CAS.
- T. Sun, P. Xu, Q. Liu, J. Xue and W. Xie, Eur. Polym. J., 2003, 39(1), 189–192 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.