
Isolation and characterization of antioxidant polysaccharides (PKCP-D70-2-a and PKCP-D70-2-b) from the Pinus koraiensis pinecone
Hua Zhangabc,
Haitian Zhaoac,
Xintao Zhouac,
Xin Yang*ac,
Siyan Shenac,
Jing Wang*ac,
Zhenyu Wangac and
Lin Gengb
aDepartment of Food Science and Engineering, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Nangang District, Harbin, 150090, China. E-mail: yangxin940@163.com; w_jing2001@126.com; Fax: +86 451 86282909; Tel: +86 45186282909
bSchool of Materials Science and Engineering, Harbin Institute of Technology, Nangang District, Harbin, 150001, China
cMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Nangang District, Harbin, 150001, China
First published on 14th November 2016
Abstract
Water-soluble polysaccharides from Pinus koraiensis pinecone were fractionated using DEAE cellulose-52 and Sephadex G-100 successively to obtain two eluents named PKCP-D70-2-a and PKCP-D70-2-b. Structural analyses showed that PKCP-D70-2-a had a higher molecular weight and degree of branching when compared to PKCP-D70-2-b. Structural analyses also showed that the monosaccharide composition of PKCP-D70-2-a differed from that of PKCP-D70-2-b. The existence of a O-glycopeptide bond in PKCP-D70-2-a and PKCP-D70-2-b was verified by a β-elimination reaction. Tertiary structure analyses indicated that PKCP-D70-2-a and PKCP-D70-2-b had a triple-helical conformation. However, PKCP-D70-2-b had more of a triple-helical conformation when compared to PKCP-D70-2-a. PKCP-D70-2-a and PKCP-D70-2-b showed stronger radical scavenging capacities. The results also signified that polysaccharides from Pinus koraiensis pinecone could be investigated as potential antioxidant agents for complementary medicine and for functional foods.
1. Introduction
Pinus koraiensis is a large conifer found mainly across Korea, Japan, and the northeastern part of China. This tree only grows in locations with altitudes higher than 1000 meters above sea level and can reach one meter in trunk diameter, and 20–30 meters in height.1 As a most important source, Pinus koraiensis is usually used in traditional Chinese medicine.2 It is used as a natural remedy for several diseases that include its effect on releasing fatigue, anti-aging, and anti-inflammatory conditions and also as an anti-neoplastic agent used to inhibit the maturation and proliferation of malignant cells.3–7 An antibacterial is similar to an antibiotic, which carries the same meaning of treatment of infections. Immunomodulation including immunopotentiation, immunosuppression, or induction of immunologic tolerance is used to adjust the immune response to a desired level. Additionally, it is used for its effect on cyclo-oxygenase activity7 and for cancer treatment and prevention.8The seeds of P. koraiensis are also used as a food supplement and the tree has been used in oriental medicine for thousands of years. The current literature testifies that the bark extract from P. koraiensis possesses antitumor, antioxidant, anti-aging, and anti-mutation activities based on removing superfluous free radicals to enhance immunity.9–11 Essential oils from the Pinus koraiensis cone show distinct evidence of antimicrobial activity and chemical composition.12 In the past, various compounds have been extracted from pinecones, such as polysaccharides, lignin-related compounds, and essential oils, among which the content of polysaccharide accounts for about 50% (w/w).13,14 Likewise, the literature reports that Xu et al. had extracted three polysaccharides fractions (PKP, PAP, and PSP) when isolated from P. koraiensis, P. armandii, and P. sylvestris var. mongolica pinecones.15 In addition, a monosaccharide composition analysis has shown that PKP, PSP, and PAP were composed mainly of Rib, Rha, Ara, Xyl, Man, Glc, and Gla in a different molar ration of heteropolysaccharides, where the PKP fractions exhibited effective scavenging activities.
For food use, food polysaccharides are preferably tasteless, odorless, colorless, and non-toxic. Therefore, to carry out rational product design and development, it was desirable to characterize the structures of these polysaccharides and to improve the understanding of the structure/property relationship. Furthermore, based on the in vitro antioxidant assays, the antioxidant capability of the polysaccharide fractions (bark, needle, and cone) from different parts of the P. koraiensis in this research was evaluated. This evaluation showed that the polysaccharide from the Pinus koraiensis pinecone (PKCP) presented a stronger scavenging ability on the 2,2-azino-bis(3-ethylbenzothiazoline-6-sulphonate) (ABTS) as well as the reducing power.
In order to develop some highly valued by-products in this study, polysaccharides were extracted from the pinecone, and several distinctive polysaccharides were further obtained. Two of them were named PKCP-D70-2-a and PKCP-D70-2-b, and were successively purified and isolated from the cone (Pinus koraiensis) using a DEAE-52 anion-exchange column and a Sephadex G-100 column successively. They were then identified through a gel permeation chromatography (GPC), a monosaccharide composition analysis, a glycopeptide bond connectivity analysis, an infrared spectroscopy, a Congo red test and finally nuclear magnetic resonance (NMR) spectroscopy assays. The results provide the first report on Pinus koraiensis pinecone polysaccharides, which can facilitate further investigation about the structure–activity relationship and health food industry application of the PKCP-D70-2-a and the PKCP-D70-2-b polysaccharides.
2. Materials and methods
2.1. Materials and reagents
At the time of their maturity during September and October 2014, the P. koraiensis cones were collected in Yichun city, Heilongjiang Province, China. After collection, the seeds were removed from the cones and the cones were cut into pieces, then oven dried at 55 °C and then crushed into powder (particle size, 250 μm, 60 mesh screen) using a Wenling DF-25 grinder (Wenling Linda Machinery Co., Ltd, Wenling, China).Folin phenol was purchased from the Tianli Chemical Reagent Co. (Tianjin, China). Coomassie brilliant blue and bovine serum albumin were both purchased from Solarbio Bioscience & Technology (Beijing, China). DEAE-52 was purchased from Whatman (Maidstone, Kent, England) and Sephadex G-100 was purchased from Pharmacia (Pharmacia Fine Chemicals, Uppsala, Sweden). The D-xylose, L-arabinose, D-glucose, D-galactose, D-mannose, D-ribose, and L-rhamnose were purchased from the Tianjin Kermel Chemical Reagent Co. (Tianjin, China). Trifluoroacetic acid (TFA) was purchased from Dikma Technologies Inc. (Beijing, China). Acetic anhydride, pyridine, acetic acid, and 2,2-azino-bis(3-ethylbenzothiazoline-6-sulphonate) (ABTS) were purchased from Sigma Co. (Harbin, China). The T-series dextrans (with bands at 194; 620; 1470; 4120; 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 840; 25820; 58400; 124, 700; 460000; 965000; and 1250000 Da) for use as molecular weight markers were purchased from Agilent (Beijing, China). All other chemicals used were of at least analytical grade.
840; 25820; 58400; 124, 700; 460000; 965000; and 1250000 Da) for use as molecular weight markers were purchased from Agilent (Beijing, China). All other chemicals used were of at least analytical grade.
2.2. Isolation, purification of PKCP and chemical characteristics analysis
The PKCP was extracted and purified according to an accepted standard deproteinization method using trichloroacetic acid (TCA)16 and depigmentation procedures with minor modifications using a DEAE-52 ion exchange column.17 Then, the polysaccharides were further separated by different concentrations of ethanol (30%, 40%, 50%, 60%, 70%, and 80%). The polysaccharides precipitation, obtained by 70% ethanol, was named PKCP-D70. A DEAE-cellulose, a column chromatography was used for purification. The crude polysaccharides (2500 mg) were then dissolved in distilled water (25 mL), applied to the column (50 mm × 100 cm), and sequentially eluted with twice the volume of the column bed of 0.05 mol L−1 NaCl. A 0.1, 0.2, 0.3, and a 0.4 mol L−1 of NaCl solution at a flow rate of 3 mL min−1 followed this procedure. Next, fractions of 20 mL eluents were collected and determined using the phenol–sulfuric acid method with D-glucose as the standard measure at 490 nm by using a T6 New Century UV/Vis spectrophotometer (Puxi Instrument Co., Ltd, Beijing, China).18 Fig. 1 reflects the elution curve with 0.1, 0.2, 0.3, and 0.4 mol L−1 of NaCl eluent. Then the PKCP-D70-2 (50 mg mL−1), as a main component, was applied to the column (25 mm × 165 cm) filled with Sephadex G-100. It was then sequentially eluted with twice the volume of the column bed using distilled water, at a flow rate of 3 mL min−1. These fractions were concentrated under reduced pressure at 55 °C and were dialyzed against distilled water using dialysis tubes with no preparation (molecular weight cutoff: 3500 Da, Mym Biological Technology Company Limited, USA) for 48 hours. The PKCP-D70-2-a and the PKCP-D70-2-b were then lyophilized. The protein contents of the purified polysaccharides were measured using the Coomassie brilliant blue method at 595 nm.19 Uronic acid was determined colorimetrically at 525 nm by a modified method described by Nelly & Gustav (1973).20 Quantitative spectrophotometric determinations of the polyphenols were accomplished using Folin phenol as the standard at 765 nm.21 The sulfuric acid group and the phosphoric acid group were determined using Qiu's method (2005).22 | ||
| Fig. 1 The elute curve of polysaccharides from the Pinus koraiensis pinecone, (a) DEAE-52 column chromatogram of D70, (b) Sephadex G-100 column chromatogram of PKCP-D70-2. | ||
2.3. Determination of monosaccharide composition
2.4. Periodate oxidation
The polysaccharide (10.0 mg) was oxidized with 0.015 mol L−1 of sodium periodate (NaIO4, 20 mL) and kept in the dark at 4 °C. Its absorbance was monitored at 223 nm every 6 h.24 The reaction was completed when the absorbance ceased descending; the formic acid production was determined by titration with 0.055 mol L−1 NaOH. The excess NaIO4 was removed by adding 2.0 mL of ethylene glycol. The solution of oxidation products was extensively dialyzed (MW cut off: 500 Da) against tap water and distilled water for 48 h, and reduced with sodium borohydride (NaBH4, 40 mg) for 12 h at 25 °C. Then, acetic acid was added to neutralize the excess NaBH4. The solution was dried, concentrated, and subjected to complete hydrolysis with 2.0 mol L−1 TFA. The acid was removed by co-distillation with MeOH under vacuum. Finally, the products were acetylated and analyzed using GC method.252.5. Carbohydrate–peptide linkage analysis
The carbohydrate–peptide linkages of the polysaccharides were analyzed by β-elimination reaction. The samples (1.0 mg mL−1, 5 mL) were incubated in 0.2 mol L−1 NaOH containing 1.0 mol L−1 NaBH4 at 45 °C for 3 h and then scanned by UV spectrophotometry from 190 to 440 nm. The resultant data was then compared with that of the alkali-untreated samples. After treatment, before and after the β-elimination reaction, each of the PKP-D70-2-a and the PKP-D70-2-b 30.0 mg amounts were detected on an automatic amino acid analyzer (Hitachi type L8800, Hitachi, Tokyo, Japan).262.6. Molecular weight determination
The purified polysaccharides (1.0 mg mL−1) were dissolved in ultrapure water; the resistivity was 18 MΩ cm at 25 °C (SIMS50000, Millipore, Boston, MA, USA). They were then characterized using gel-permeation chromatography (GPC) (Agilent 1100, Agilent Technologies, Santa Clara, CA, USA). The instrument was equipped with an Agilent Technologies PL Aquagel-OH mixed column (300 mm × 7.5 mm i.d. × 8 μm) and an Agilent G1362A refractive index detector. The samples were then injected into the column using an Agilent G1313A Autosampler and were then eluted with 0.02% NaN3 in ultrapure water at 25 °C at a flow rate of 1.0 mL min−1.272.7. UV analysis
A UV scan can identify homogeneous components of nucleic acid and protein in polysaccharides. In this study, polysaccharide solution (0.1 mg mL−1) was analyzed by ultraviolet scanning in the range of 190–400 nm.282.8. FTIR analysis
The structural characteristics of the polysaccharide samples were recorded on a Spectrum One B, Fourier transform infrared spectrophotometer (PerkinElmer Inc., Waltham, MA, USA); and then operated in the 4000–400 cm−1 region at a nominal resolution of 4 cm−1, taking 32 scans for each sample. The samples were prepared by grinding the polysaccharides with dry KBr and then pressing the samples into pellets prior to running.292.9. Congo red test
According to the method of Satitmanwiwat with some slight modifications, each polysaccharide (5.0 mg) was added to 2.0 mL of distilled water and 2.0 mL of Congo red reagent.30 Different volumes of 1.0 mol L−1 NaOH solution were then added to obtain a final concentration from 0 mol L−1, which was gradually increased to 0.4 mol L−1 (0, 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, and 0.4 mol L−1) (Satitmanwiwat et al., 2012). The samples were mixed and scanned in the visible wavelength range of 400 to 600 nm. The maximum absorption wavelengths of the sample solution under different alkaline conditions were recorded.312.10. NMR spectroscopy
Spin systems in the polysaccharides and their sequential assignments were identified by recording NMR spectra on a 500 MHz Bruker Avance III spectrometer (Bruker, Stockholm, Sweden). Up to 20.0 mg of each of the samples were separately collected into two NMR tubes and then separately dissolved in 0.5 mL of D2O at 25 °C. The spectra were recorded at 353 K on a Bruker Avance III spectrometer operating at 500 MHz for 1H and 125.75 MHz for 13C. 1H NMR and 13C NMR spectra were measured using a superconducting magnetic resonance instrument.32 The carbon spectrum signal acquisition time was 15 h.2.11. Antioxidant activity assay
The scavenging ability was calculated as follows:
| Scavenging ability (%) = [1 − (A1 − A2)/A0] × 100% | (1) |
|
Scavenging ability% = [1 − (A1 − A2)/A0] × 100%
| (2) |
The reducing power was calculated as follows:
| Reducing power (%) = A1 − A0 | (3) |
2.12. Statistical analysis
The data are presented as means ± standard deviations of three determinations. Statistical analyses were done using a Student's t-test and a One-way Analysis of Variance (ANOVA). All computations were done using statistical software SPSS 16.0 (R package). p ≤ 0.05 was considered statistically significant.3. Results and discussion
3.1. Extraction, purification and physicochemical properties of polysaccharides
After removing the pinecone seeds, together with the protein and pigmentation, a water-soluble polysaccharide was obtained from the Pinus koraiensis pinecones and was named PKCP. The PKCP was then graded with an alcohol precipitation with 70% ethanol and it was named as PKCP-D70. The physicochemical properties of the PKCP and PKCP-D70 are illustrated in Table 1. Then, following isolation and purification by the DEAE cellulose-52 and the Sephadex G-100, the PKCP-D70-2-a and the PKCP-D70-2-b were obtained from the PKCP-D70 (the elute curves are shown in Fig. 1a and b). The yields of the PKCP-D70-2-a and the PKCP-D70-2-b were approximately 43% and 47%. Their physicochemical properties are illustrated in Table 2. The antioxidant properties of the polysaccharides correlated with the ratios of different monosaccharides and the content of total sugars, uronic acid, Pro, and polyphenols. The total sugar content and the uronic acid for the PKCP-D70-2-a and PKCP-D70-2-b were 74.8% and 80%, respectively.| Component (%) | Polysaccharides | |
|---|---|---|
| PKCP | PKCP-D70 | |
| a Values within a row with different letters are significantly different at p ≤ 0.05. | ||
| Total sugar | 42.7 ± 0.4b | 58.3 ± 0.5a |
| Uronic acid | 52.6 ± 0.7a | 40.0 ± 0.4b |
| Protein | 1.2 ± 0.1a | 0.5 ± 0.1b |
| Polyphenol | 3.0 ± 0.5a | 1.2 ± 0.1b |
| Component (%) | Polysaccharide | |
|---|---|---|
| PKCP-D70-2-a | PKCP-D70-2-b | |
| a Values within a row with different letters are significantly different at p ≤ 0.05. | ||
| Total sugar | 52.7 ± 0.5a | 34.7 ± 0.4b |
| Uronic acid | 22.1 ± 0.4b | 45.3 ± 0.3a |
| Protein | 0.32 ± 0.03a | 0.28 ± 0.05a |
| Polyphenol | 0.89 ± 0.09a | 0.92 ± 0.10a |
| Sulfuric acid group | 2.25 ± 0.10a | 2.06 ± 0.20a |
| Phosphoric acid group | 0.114 ± 0.012a | 0.165 ± 0.013a |
3.2. Molecular weight distribution
A typical size-exclusion GPC chromatogram of the PKCP-D70-2-a and the PKCP-D70-2-b peaked in the region of 0–14 min (Fig. 2), which confirmed that the PKCP-D70-2-a and the PKCP-D70-2-b were both homogeneous samples. The molecular weights of PKCP-D70-2-a and PKCP-D70-2-b were 3048 and 2548 kDa, respectively. The molecular weight of polysaccharide has a significant effect on solution viscosity. As the molecular weight increases, the viscosity of the polymer solution also increases. Moreover, polysaccharides with relative high molecular weight have a greater advantage in stimulating macrophage activity.36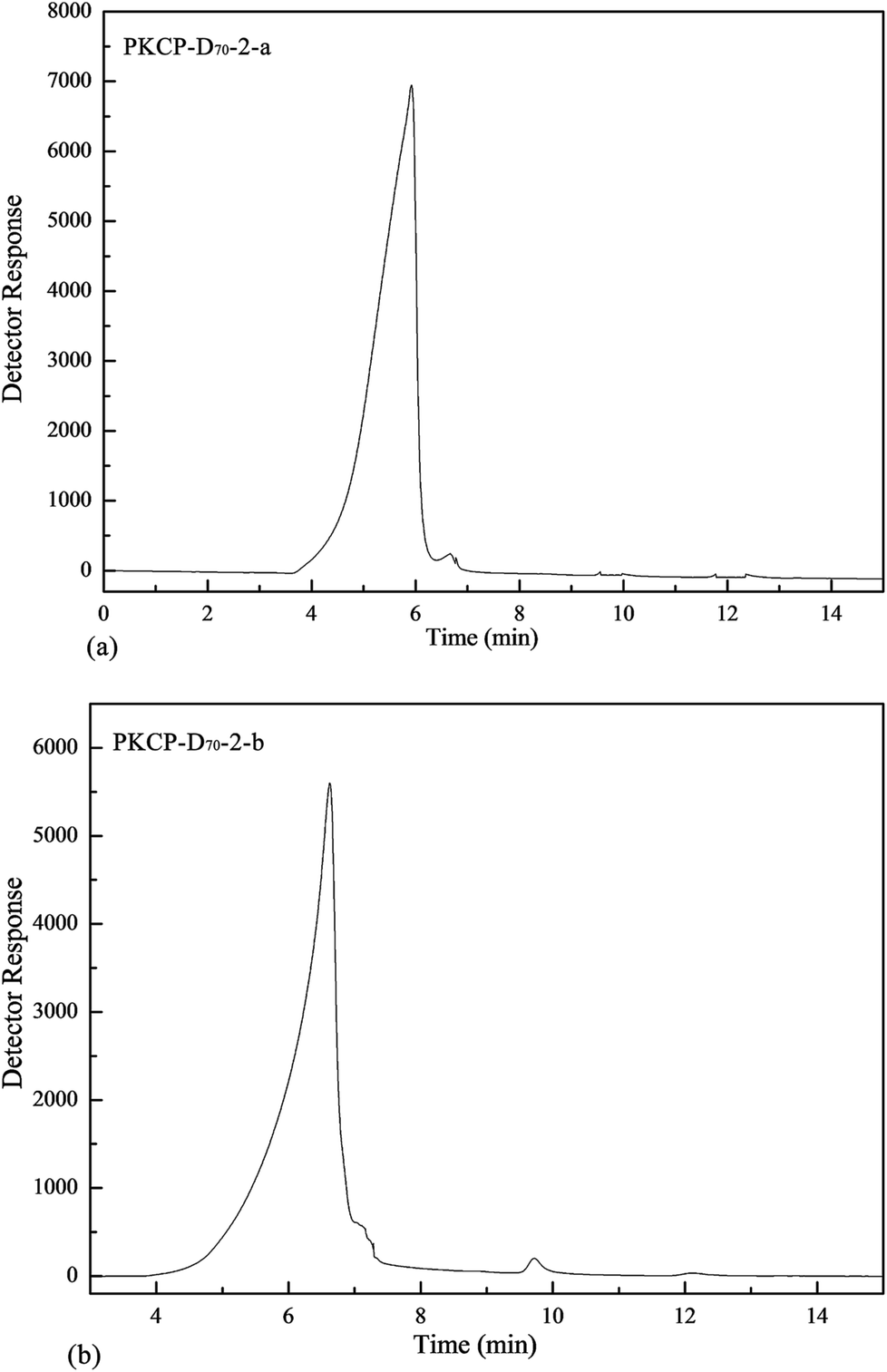 | ||
| Fig. 2 Molecular weight distributions of PKCP-D70-2-a and PKCP-D70-2-b. | ||
3.3. Monosaccharide composition
To improve an understanding of the structure/property relationship and to conduct a rational product design and development necessitated, the characterization of the structures of the polysaccharides was illustrated. Therefore, a complete acid hydrolysis and a partial acid hydrolysis were employed to determine the monosaccharide composition of both the main and side chains. Subsequently, the released monosaccharide derivatives (alditol acetate approach) were analyzed under optimized conditions by the GC-MS method described above.As illustrated in Fig. 3b and c, the composition of the PKCP-D70-2-a and the PKCP-D70-2-b were identified. The heteropolysaccharides were correspondingly composed of Rha, Ara, Man, Glc and Gal with a molar ratio of 13.6:10.7:20.2:19.2:36.3 and Ara, Man, Glc and Gal with a molar ratio of 13.7:36.0:27.2:23.1, respectively.
 | ||
| Fig. 3 Gas chromatogram of monosaccharide standard, PKP-D70-2-a and PKCP-D70-2-b, (a) gas chromatogram of monosaccharide standard, (b) gas chromatogram of PKP-D70-2-a, (c) gas chromatogram of PKCP-D70-2-b. | ||
The results of the partial acid hydrolysis (shown in Table 3) specified that the main chain of the PKCP-D70-2-a contained four monosaccharides, including L-rhamnose, D-mannose, D-glucose, and D-galactose with D-mannose occupying the maximum molar ratio of 50.46% in the main chain; then followed by D-galactose, D-glucose, and L-rhamnose. However, in the branched chain of the PKCP-D70-2-a, there were five monosaccharides, including D-galactose, D-mannose, D-glucose, L-rhamnose, and L-arabinose. The content of the five monosaccharides successively decreased. The branched degree in the branched chain of the PKCP-D70-2-a reached 55.49%. From the above results, L-arabinose only exists in the presence of the PKCP-D70-2-a branch. Alternatively, confirmed by acid hydrolysis, the proportion was the least with the key monosaccharides in the main and branched chain D-galactose and D-mannose.
| Monosaccharide | Retention time (min) | Molar ratio | |||||
|---|---|---|---|---|---|---|---|
| Fraction 1 | Fraction 2 | Fraction 3 | |||||
| PKCP-D70-2-a | PKCP-D70-2-b | PKCP-D70-2-a | PKCP-D70-2-b | PKCP-D70-2-a | PKCP-D70-2-b | ||
| a Note: N.D—not detected. | |||||||
| L-Rhamnose | 6.14 | 10.91 | N.D | 4.20 | N.D | 6.19 | N.D |
| L-Arabinose | 6.21 | 11.24 | 8.32 | 2.46 | 3.66 | N.D | 10.18 |
| D-Mannose | 8.40 | 35.85 | 64.16 | 28.59 | 39.05 | 50.46 | 36.23 |
| D-Glucose | 8.50 | 23.62 | 15.51 | 15.75 | 29.66 | 19.78 | 29.03 |
| D-Galactose | 8.60 | 18.38 | 12.01 | 49.00 | 30.63 | 23.56 | 24.66 |
The stability of the main and branched chain of polysaccharide differed. In general, different concentrations of TFA are regularly used to hydrolyze glycosides in polysaccharide. Consequently, the monosaccharide compositions of the main and branched chain of the polysaccharide were ascertained. The results of the partial acid hydrolysis showed that the main chain of the PKCP-D70-2-b contained four monosaccharides, with D-mannose occupying the molar ratio of a maximum 36.23% in the main chain; then followed by D-glucose, D-galactose, and L-rhamnose (shown in Table 3). The branched degree in the branched chain of the PKCP-D70-2-b was 53.80%. From the above data, the main type of monosaccharides in the main and branched chain was confirmed by acid hydrolysis.
3.4. Periodate oxidation
Sarkar (1976) used the periodate oxidation reaction process followed by methylation studies of polysaccharide, to determine the structure of seed polysaccharide.37 The polysaccharide contained free hydroxyl groups resulting in the consumption of periodate-ions during the periodate oxidation reaction. Formic acid from the reducing and non-reducing terminal unit of the D-glucose unit indicated that the polysaccharide had 1→6 or 1→glycosides. In our research, the structure of the PKCP-D70-2-a and the PKCP-D70-2-b, obtained after methylation studies, was confirmed by the periodate oxidation results. Following Smith degradation, the periodate oxidation products of the PKCP-D70-2-a consumed 0.038 mmol of periodate and liberated 0.0028 mmol of formic acid per mol of anhydrohexose sugar units. This outcome specified different kinds of glycosidic bonds existed in the PKCP-D70-2-a, including 1→2 or 1→2,6, 1→4, or 1→4,6. Likewise, the molar proportion of 1→6 or 1→linked glycosyl glycosides of the PKCP-D70-2-a attained 14.74%. Other connected residues such as 1→3, 1→3,6, 1→2,3, 1→2,4, 1→3,4, 1→2,3,4 were not periodate oxidation. Among them, by periodate oxidation of carbohydrate residues, were 1→2 or 1 →2,6 and 1→4 or 1→4,6, which accounted for a molar ratio of 85.26%.Smith degradation characteristics are glycosidic bonds oxidized by periodate; they are only broken while sugar residues are even on the sugar chain without exhibiting periodate oxidation. Table 4 illustrates that a large amount of glycerol and erythritol appeared in the PKCP-D70-2-a product. To illustrate, the resulting glycerol residues and glycosidic bond type: 1→, 1→6, 1→2, 1→2,6, and the erythritol residue glycosidic bond type: 1→4, 1→4,6, were consistent with the Smith degradation principle diagram.
| Monosaccharide | Retention (min) | Smith degradation molar ratio | |
|---|---|---|---|
| PKCP-D70-2-a | PKCP-D70-2-b | ||
| a Note: N.D—not detected. | |||
| Glycerol | 4.19 | 16.95 | 13.73 |
| Erythritol | 4.91 | 17.68 | 48.5 |
| L-Rhamnose | 6.14 | 1.46 | N.D |
| L-Arabinose | 6.21 | 4.45 | 2.62 |
| D-Mannose | 8.40 | 14.95 | 4.41 |
| D-Glucose | 8.50 | 27.83 | 6.41 |
| D-Galactose | 8.60 | 16.83 | 24.29 |
L-Rhamnose, L-arabinose, D-mannose, D-glucose and D-galactose were also detected in the PKCP-D70-2-a degradation products and were linked with (1→3), (1→3,6), (1→2,3), (1→2,4), (1→3,4) or (1→2,3,4) glycosidic bonds, which incapable of being oxidized by periodate. This result showed a good correlation between terminal and branched residues. In addition, the molar ratios also agreed with these results together with the overall monosaccharide composition of the PKCP-D70-2-a described above.
A comparison with the Smith degradation of PKCP-D70-2-a, found that the difference was only four monosaccharides without L-rhamnose detected in the PKCP-D70-2-b degradation products. Illustrated in main glycosidic bond configuration in PKCP-D70-2-a and PKCP-D70-2-b characterization by 1H NMR and 13C NMR, each was linked with (1→3), (1→3,6), (1→2,3), (1→2,4), (1→3,4) or (1→2,3,4) glycosidic bonds, which were not capable of being oxidized by the periodate.
3.5. Congo red test
A Congo red test is an acid dye used to form triple helix structures. When the Congo red test was used in this research the maximum absorption wavelength of the complex with polysaccharides changed.38 With increasing concentrations of NaOH, the maximum absorption wavelength of the complex, when blended with PKCP-D70-2-a, did not shift to “long wave” in the presence of the Congo red to any significance when compared with the Congo red test of the PKCP-D70-2-b (see Fig. 4). This result confirmed that the PKCP-D70-2-a had fewer helical configurations in common with a free-coiled conformation. Conversely, the maximum absorption wavelength of the PKCP-D70-2-b complex significantly shifted to “long wave” when compared with the Congo red test conducted on the PKCP-D70-2-a. Fig. 4 shows that the maximum absorption wavelength continuously decreased, which was most likely caused by the remaining hydrogen bonds of the PKCP-D70-2-b being destroyed by the presence of sodium hydroxide increasing range from 0.2 to 0.4 mol L−1. As the helical structure of polysaccharide has a certain relationship with its functionality, the results confirmed that the PKCP-D70-2-a and the PKCP-D70-2-b exhibited a triple helix structure, whereas the PKCP-D70-2-b offered considerable helical configuration structures. | ||
| Fig. 4 Helix–coil transition analysis of the isolated polysaccharides (PKCP-D70-2-a and PKCP-D70-2-b) at different concentration of NaOH. | ||
3.6. Carbohydrate–peptide linkage analysis
A β-elimination reaction is mainly used to determine the type of linkage with O-glycosides between a polysaccharide and the amino acid in glycoprotein. N-Glycosides and O-glycosides are the two main types of glycosides that exist between polysaccharide and amino acid. O-Glycosides form when beta hydroxyl of Ser or Tyr is combined with glycosyl. O-Glycosides are unstable in an alkaline condition. Therefore, in the β-elimination reaction, a certain relationship existed between the increase of α-alanine and aminobutyric acid and the decrease of serine (Ser) and threonine (Thr).39 In this research, after alkaline hydrolysis, the UV scanning spectrum solution, with an absorbance set at 240 nm of PKCP-D70-2-a and PKCP-D70-2-b, significantly increased. This result showed that these polysaccharides exhibited O-glycosidic bonds (Fig. 5).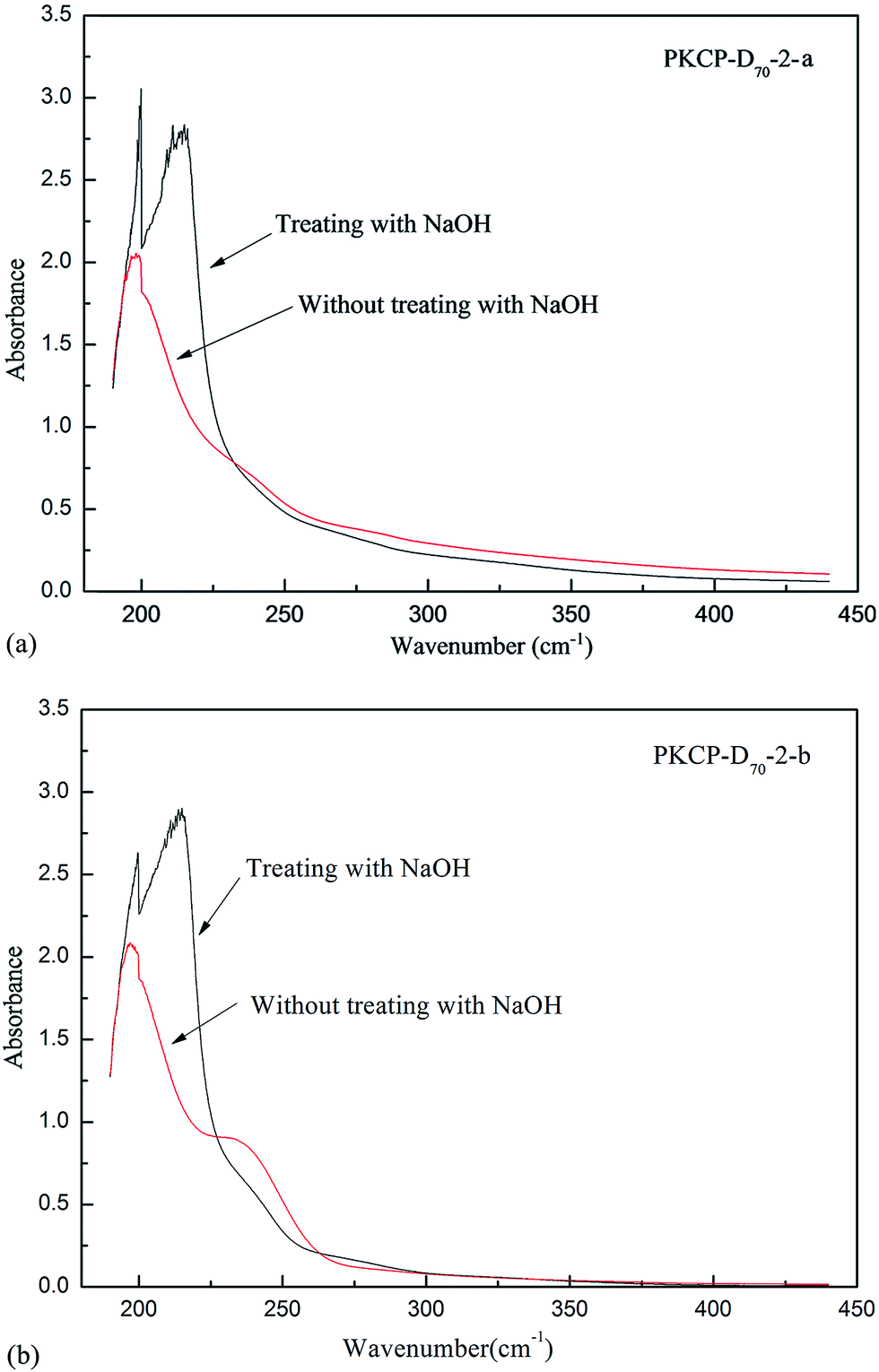 | ||
| Fig. 5 The result of PKP-D70-2-a and PKCP-D70-2-b by β-elimination reaction. | ||
In addition, the tests showed that the sodium borohydride (1.0 mol L−1, 5 mL) was concluded in the β-elimination reaction system, α-acrylic acid was reduced to alanine, and α-amino crotonic acid was reduced to α-amino butyric acid. Consequently, a relationship subsequently existed between the increase of α-alanine and aminobutyric acid and the decrease of serine and threonine in the β-elimination reaction.
Tables 5 and 6 show that the contents of several important amino acids in PKCP-D70-2-a and PKCP-D70-2-b changed for β-elimination reaction, up to 0.03 nmol of asparagine and 0.08 nmol of serine increased with the loss of 1.01 nmol of threonine per nmol of the PKCP-D70-2-a; with the loss of 0.070 nmol of serine and 0.002 nmol threonine per mol of the PKCP-D70-2-b. Content changes in the amino acids of the two polysaccharides revealed the PKCP-D70-2-a exhibited O-peptide bond (–O-Thr) connections and the PKCP-D70-2-b exhibited O-peptide bond (–O-Ser or –O-Thr) connections and mainly contained O-Ser.12
| Amino acids | Before β-elimination reaction | After β-elimination reaction | ||
|---|---|---|---|---|
| Content (ng) | Molar ratio (nmol) | Content (ng) | Molar ratio (nmol) | |
| a Note: N.D—not detected. | ||||
| Asp | N.D | N.D | 0.17 | 0.03 |
| Thr | 5.36 | 1.07 | 0.36 | 0.06 |
| Ser | N.D | N.D | 0.41 | 0.08 |
| Glu | N.D | N.D | 0.69 | 0.09 |
| Gly | 1.04 | 0.33 | 0.47 | 0.12 |
| Cys | 1.20 | 0.24 | 2.07 | 0.35 |
| Val | 5.24 | 0.53 | 3.37 | 0.28 |
| Met | 0.18 | 0.03 | 3.66 | 0.48 |
| Ile | 0.07 | 0.01 | 0.81 | 0.12 |
| Leu | 0.21 | 0.04 | 0.66 | 0.10 |
| Tyr | 0.14 | 0.02 | 0.17 | 0.02 |
| His | 0.13 | 0.02 | N.D | N.D |
| Arg | 0.03 | 0.00414 | 0.03 | 0.00371 |
| Amino acids | Before β-elimination reaction | After β-elimination reaction | ||
|---|---|---|---|---|
| Content (ng) | Molar ratio (nmol) | Content (ng) | Molar ratio (nmol) | |
| a Note: N.D—not detected. | ||||
| Asp | 0.08 | 0.02 | 0.16 | 0.03 |
| Thr | 0.31 | 0.06 | 0.30 | 0.06 |
| Ser | 0.72 | 0.16 | 0.42 | 0.09 |
| Glu | N.D | N.D | 1.13 | 0.18 |
| Gly | 0.07 | 0.02 | 0.09 | 0.03 |
| Cys | 1.42 | 0.29 | 2.05 | 0.41 |
| Val | 2.24 | 0.22 | 3.64 | 0.35 |
| Met | 3.49 | 0.56 | 0.24 | 0.04 |
| Ile | N.D | N.D | 0.25 | 0.05 |
| Leu | 0.82 | 0.15 | N.D | N.D |
| Tyr | N.D | N.D | N.D | N.D |
| His | 0.13 | 0.02 | N.D | N.D |
| Arg | 0.05 | 0.01 | 0.03 | 0.003 |
3.7. UV analysis
The UV absorption wavelength of the proteins and the nucleic acids ranged from 260–280 nm; consequently, this absorption peak was used to determine whether or not the polysaccharide solution contained proteins and nucleic acids.Both the PKCP-D70-2-a and the PKCP-D70-2-b were scanned at a wavelength of 190–400 nm. As shown in Fig. 6, the PKCP-D70-2-a and the PKCP-D70-2-b did not contain nucleic acid or tryptophan. In addition, the protein content in the PKCP-D70-2-a and the PKCP-D70-2-b was so low that there was no obvious peak at 280 nm and the protein content was absent in both the UV spectrum of the PKCP-D70-2-a and the PKCP-D70-2-b. This outcome was consistent with results of the chemical composition analysis.40
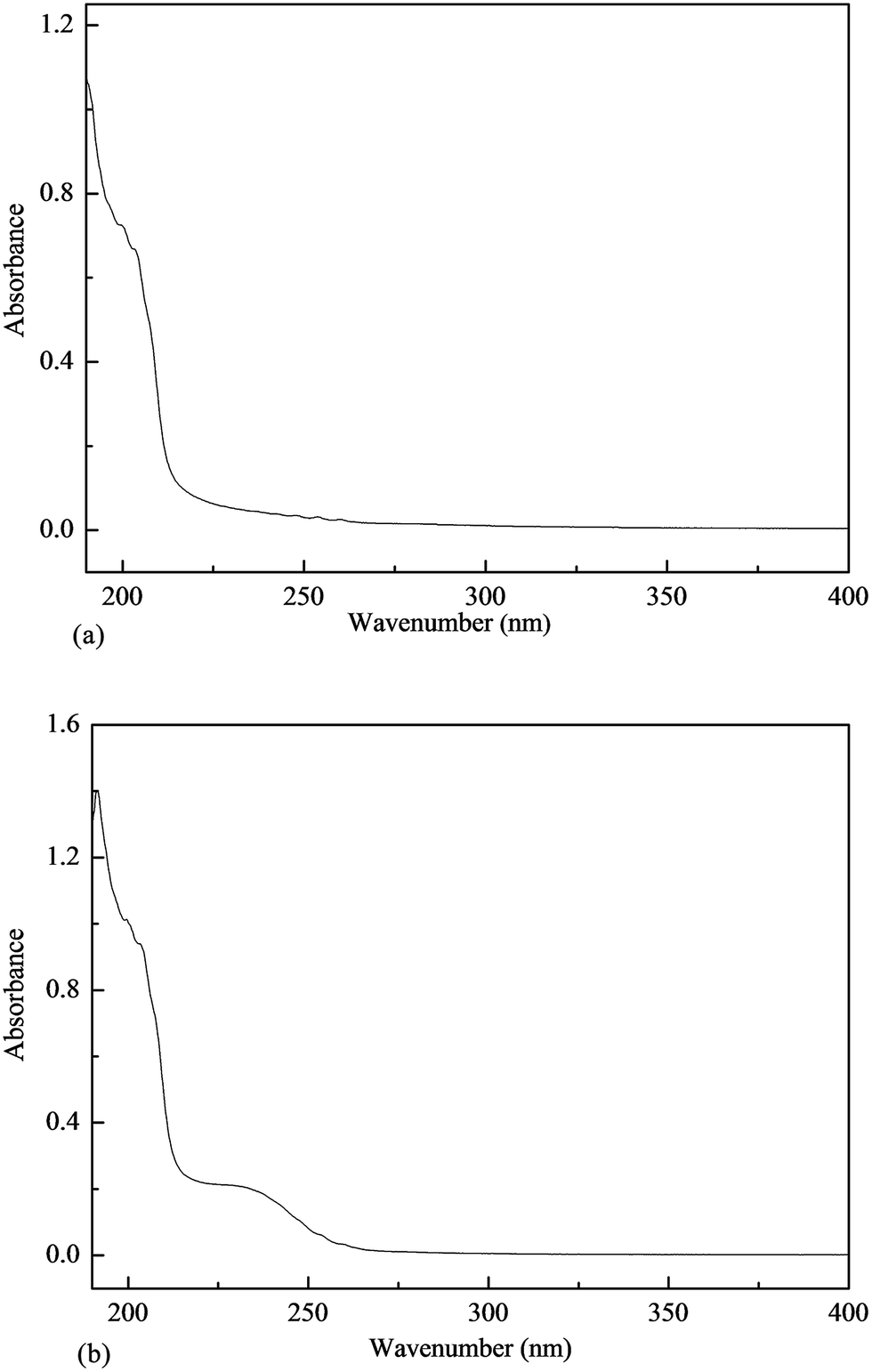 | ||
| Fig. 6 UV spectra of PKCP-D70-2-a and PKCP-D70-2-b, (a) PKCP-D70-2-a, (b) PKCP-D70-2-b. | ||
3.8. FTIR analysis
A FTIR spectra analysis (Fig. 7) was used to provide further structural information in relation to the PKCP-D70-2-a and the PKCP-D70-2-b. Different absorption bands for the FTIR analysis were assigned as previously described. In the 3400, 3429 and the 2927, 2941 cm−1 of each of the two polysaccharides, the infrared spectra were attributed to the OH stretching vibrations in the hydrogen bonds and the C–H stretching vibrations (Fig. 7). In the 1736, 1747 and 1611, 1615 cm−1 range of the two polysaccharides, the infrared spectra were ascribed to the absorption of the COO− deprotonated carboxylic group.41 The absorption peaked at 1073 and 1102 cm−1 to indicate the presence of a β-pyran linkage.42 The PKCP-D70-2-b absorption peaked at 832 cm−1 to indicate α-glycopyranosidic linkages, while the absorption peak at 830 cm−1 (830–836 cm−1) suggested the presence of α-glycosidic bonds.43 The absorption peaks of 1385 and 1420 cm−1, caused by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetry vibration, showed the presence of polyphenols in both the PKCP-D70-2-a and the PKCP-D70-2-b, which was consistent with results of the chemical composition analysis.
O symmetry vibration, showed the presence of polyphenols in both the PKCP-D70-2-a and the PKCP-D70-2-b, which was consistent with results of the chemical composition analysis.
 | ||
| Fig. 7 FTIR spectra of PKCP-D70-2-a and PKCP-D70-2-b, (a) PKCP-D70-2-a, (b) PKCP-D70-2-b. | ||
The absorption peak of 1255 and 1237 cm−1 also illustrated an asymmetrical SO stretching vibration, showing the presence of sulfate groups in both the PKCP-D70-2-a and the PKCP-D70-2-b. However, the PKCP-D70-2-b may offer considerable sulfate groups. The C–OH and the C–O–C stretching vibration caused a characteristic absorption at 1200–1000 cm−1 in the PKCP-D70-2-b. Only the PKCP-D70-2-b showed a weak absorption peak at 800–850 cm−1. This outcome implied that the PKCP-D70-2-b exhibited sulfate groups, while the absorption peaked at 832 cm−1, and attributed to α-configurations. Overall, both the PKCP-D70-2-a and the PKCP-D70-2-b displayed both β-D-pyran-type and α-D-pyran-type sugar rings.44 It was concluded that the polysaccharide molecules presented considerable linkage to the β-type glycosidic bond.
3.9. NMR spectroscopy
NMR spectroscopy of 1H and 13C is an efficient method to analyze the structural features of polysaccharide.45,46 The 1H-NMR spectrum showed that the other proton signal was assigned the α-D-galacturonic acid (GaluA) H-1 at 5.05 ppm.47,48 The chemical shifts from 0.9 to 4.7 ppm (Fig. 8a), showing the overlapping peaks, were assigned to protons of carbons C-2 to C-5 (or C-6) of the glycosidic ring. The chemical shift of anomeric region at δ 4.9 ppm in PKCP-D70-2-a is less than that of δ 5.0 ppm and can be demonstrated by β-type configurations, while the chemical shift of anomeric region at δ 5.0 and δ 5.1 ppm is greater than that of δ 5.0 ppm to demonstrate α-type configurations.49 In addition, the chemical shift of anomeric region at δ 4.3 ppm, δ 4.6 ppm and δ 4.8 is less than that of δ 5.0 ppm. This outcome confirmed that the PKCP-D70-2-b was a β-type configuration, while the chemical shift of anomeric region at δ 5.0 and δ 5.4 ppm was greater than that of δ 5.0 ppm to confirm that the PKCP-D70-2-b was also a α-type configuration.49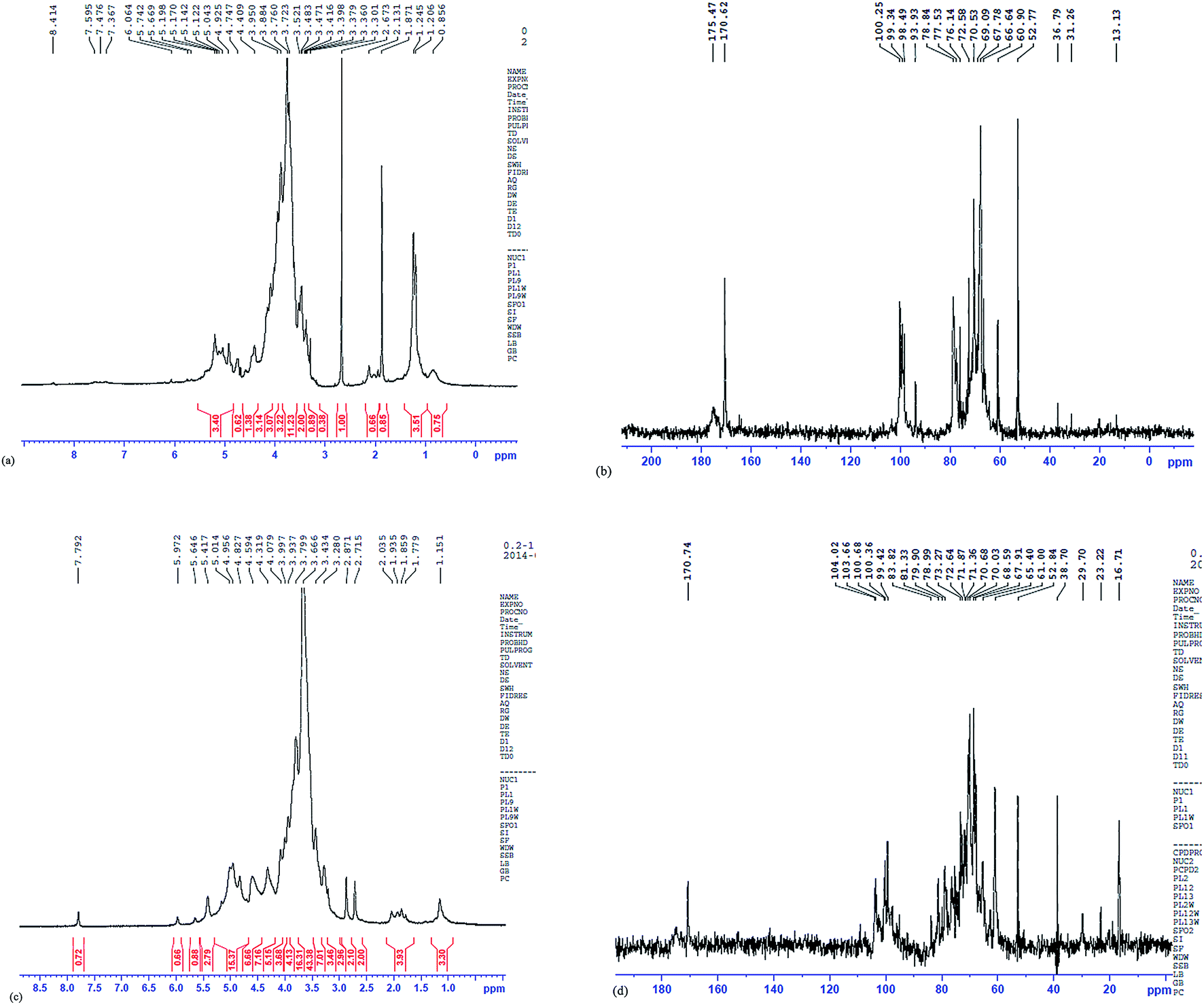 | ||
| Fig. 8 1H-NMR and 13C-NMR of PKP-D70-2-a and PKCP-D70-2-b, (a) 1H-NMR of PKCP-D70-2-a, (b) 13C-NMR of PKCP-D70-2-a, (c) 1H-NMR of PKCP-D70-2-b, (d) 13C-NMR of PKCP-D70-2-b. | ||
Correlations between the anomeric protons and the transglycosidic carbons were observed in the 13C-NMR of PKCP-D70-2-a and PKCP-D70-2-b (Fig. 8b and d). This outcome confirmed the monosaccharide composition of the PKCP-D70-2-a and PKCP-D70-2-b, and the attachment sites. In the 13C NMR spectrum, the signal at δ 100.2 ppm of PKCP-D70-2-a was reasonably assigned to the unsubstituted →4)-α-D-Galp-(1→ of terminal residue A; while, the signal at δ 104.0 ppm of PKCP-D70-2-b was reasonably assigned to the unsubstituted β-D-Glcp-(1→. The C-1 signal of the 3,4)-a-Galp-(1→)-linked residue C appeared at δ 99.3 ppm of PKCP-D70-2-a. The C-1 signal of 4,6)-β-Glcp-(1→)-linked residue C appeared at δ 98.5 ppm of PKCP-D70-2-a and α-D-Gla-(1→6) appeared at δ 99.4 ppm of the PKCP-D70-2-b, which originated from C-1, C-2, C-3, C-4 and C-5.50 Given the signals at δ 70.0 to δ 78.0 that densely overlapped and combined with the monosaccharide composition analysis, the PKCP-D70-2-a was considered a heterosaccharide. The shift of C2–C6 from 52.8 to 78.8 ppm, together with the signal at δ 170.6 ppm was due to the CO of the esterification of α-D-galacturonic acid, δ 175.5 ppm. This result was attributed to the CO for the non-esterification of the α-D-galacturonic acid,51 which accounts for the large volume of uronic acid in the composition and is consistent with the chemical composition analysis. The NMR spectral analysis was confirmed when supported by the conclusions drawn from the GLC and FTIR data.
The relevant literature analysis,52–55 and the signals in 1H-NMR and the 13C-NMR of PKCP-D70-2-a and PKCP-D70-2-b are correspondingly illustrated in Tables 7–10.
| Structure | 1H chemical shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|
| H-1 | H-2 | H-3 | H-4 | H-5 | H-6 | ||
| 3,4)-α-Galp-(1→2) | Literature value | 5.290 | 4.111 | 4.091 | 4.323 | 4.134 | 3.66 |
| Observed value | 5.198 | — | 3.950 | 4.409 | — | 3.723 | |
| →4)-α-D-Galp-(1→ | Literature value | 4.983 | 3.634 | 3.852 | 4.344 | 4.953 | — |
| Observed value | 5.043 | 3.723 | 3.884 | 4.409 | 4.925 | — | |
| 4,6)-β-Glcp-(1→4) | Literature value | 4.941 | 3.946 | 3.946 | 3.988 | 4.504 | 4.230 |
| Observed value | 4.925 | 3.950 | 3.950 | 3.950 | 4.409 | — | |
| Structure | 13C chemical shift (ppm) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | O/N-Ac | O-Ac | ||
| 3,4)-a-Galp-(1→2) | Literature value | 99.7 | 69.1 | 69.8 | 79.3 | 71.3 | 171.8 | 21.3 | 21.4 |
| Observed value | 99.34 | 69.09 | 70.53 | 78.84 | — | 170.62 | 174.4 | 174.3 | |
| →4)-α-D-Galp-(1→ | Literature value | 100.1 | 69.1 | 79.3 | 77.1 | 73.1 | 61.0 | ||
| Observed value | 100.25 | 69.09 | 78.84 | 77.53 | 72.58 | 60.90 | |||
| 4,6)-β-Glcp-(1→4) | Literature value | 98.0 | 54.7 | 70.1 | 78.7 | 70.1 | 67.9 | ||
| Observed value | 98.49 | — | 70.53 | 78.84 | 70.53 | 67.78 | |||
| Structure | Chemical shift | ||||||
|---|---|---|---|---|---|---|---|
| H-1 | H-2 | H-3 | H-4 | H-5 | H-6 | ||
| β-D-Glcp-(1→ | Literature value | 4.82 | 3.38 | 3.43 | 3.41 | 3.35 | 3.75 |
| Observed value | 4.827 | 3.280 | 3.438 | 3.434 | — | 3.779 | |
| β-D-Gal link α-D-Gal-6 sulfate | Literature value | 4.43 | 3.72 | — | — | — | 3.51 |
| Observed value | 4.319 | 3.79 | — | — | — | 3.43 | |
| →4)-β-D-GalpA6Me-(→ | Literature value | 4.794 | 3.624 | 3.849 | 4.293 | 4.527 | — |
| Observed value | 4.594 | 3.666 | 3.799 | — | — | — | |
| →4)-α-D-Glc-(1→ | Literature value | 5.06 | 3.62 | 3.87 | 3.82 | 3.89 | 3.55 |
| Observed value | 5.014 | 3.66 | 3.937 | 3.799 | 3.997 | 3.666 | |
| α-D-Glc-(1→) | Literature value | 5.31 | 3.55 | 3.45 | 3.98 | 3.63 | 3.65 |
| Observed value | 5.41 | 3.434 | 3.434 | 3.997 | 3.666 | 3.700 | |
| Structure | Chemical shift | ||||||
|---|---|---|---|---|---|---|---|
| C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | ||
| β-D-Glcp-(1→ | Literature value | 103.93 | 74.85 | 77.55 | 71.15 | 77.18 | 62.25 |
| Observed value | 104.02 | — | 78.99 | 71.36 | 78.99 | — | |
| β-D-Gal link α-D-Gal-6 sulfate | Literature value | 103.7 | 70.8 | 80.3 | 69.2 | 76.0 | 61.9 |
| Observed value | 103.66 | 70.63 | 79.90 | 68.59 | — | 61.00 | |
| →4)-β-D-GalpA6Me-(→ | Literature value | 100.70 | 69.10 | 69.80 | 79.3 | 72.3 | — |
| Observed value | 100.68 | 68.59 | 70.03 | 79.90 | 72.64 | — | |
| →4)-α-D-Glc-(1→ | Literature value | 100.40 | 72.36 | 74.88 | 70.50 | 66.05 | 62.13 |
| Observed value | 100.36 | 72.64 | — | 70.68 | 65.40 | 61.00 | |
| α-D-Gla-(1→ | Literature value | 99.64 | 71.39 | 73.68 | 69.31 | 69.43 | 60.07 |
| Observed value | 99.42 | 71.36 | 73.27 | 68.59 | 70.03 | 61.00 | |
The relatively downfield carbon chemical shifts at δ 60–79 ppm, caused by the α-glycosylation effect, were assigned to substitute C2–C6 together with the signal at δ 170.6 ppm. Due to the CO of the esterification of α-D-galacturonic acid, δ 175.5 ppm was attributed to CO and for the non-esterification of α-D-galacturonic acid.51 This outcome shows that within this composition there was a large volume of uronic acid, which is consistent with the current chemical composition analysis, was confirmed by the NMR spectral analysis, and was further supported by the conclusions drawn from the GLC and FTIR data. Given that no anomeric proton existed at δ 5.4, the PKCP-D70-2-b had pyran-type sugar rings. These findings were consistent with an infrared spectrum analysis. In conclusion, the PKCP-D70-2-b had a backbone chain mainly composed of β-D-Glcp-(1→, β-D-Gal link α-D-Gal-6 sulfate, →4)-β-D-GalpA6Me-(→, →4)-α-D-Glc-(1→, α-D-Glc-(1→), and also showed other glycosidic bond connections, which required further exploration. To conclude, the PKCP-D70-2-a had a backbone chain mainly composed of three glycosidic bonds 3,4)-α-Galp-(1→), →4)-α-D-Galp-(1→ and 4,6)-β-Glcp-(1→4), and presented other glycosidic bond connections, which required further exploration.
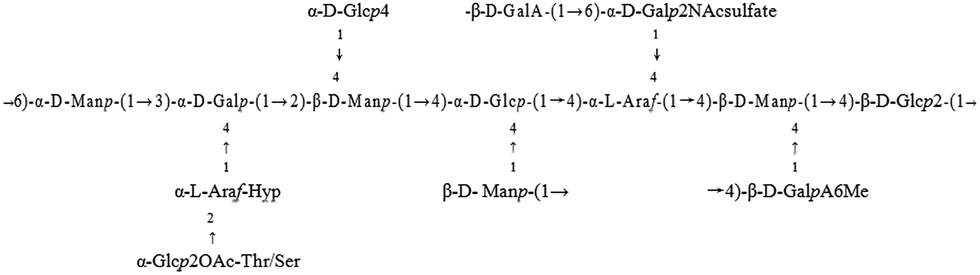
For better elucidation of PKCP-D70-2-a and PKCP-D70-2-b structures using techniques e.g., hydrolysis (complete and partial acid hydrolysis), methylation, acetylation, periodate oxidation, carbohydrate–peptide linkage analysis, FTIR analysis, GC/MS, 1H-NMR and 13C-NMR, instead of 2D NMR. Part of structural features of the PKCP-D70-2-a and the PKCP-D70-2-b could be predicted as followed:
Structural features of the PKCP-D70-2-a:

Structural features of the PKCP-D70-2-b:
3.10. Antioxidant activity assay
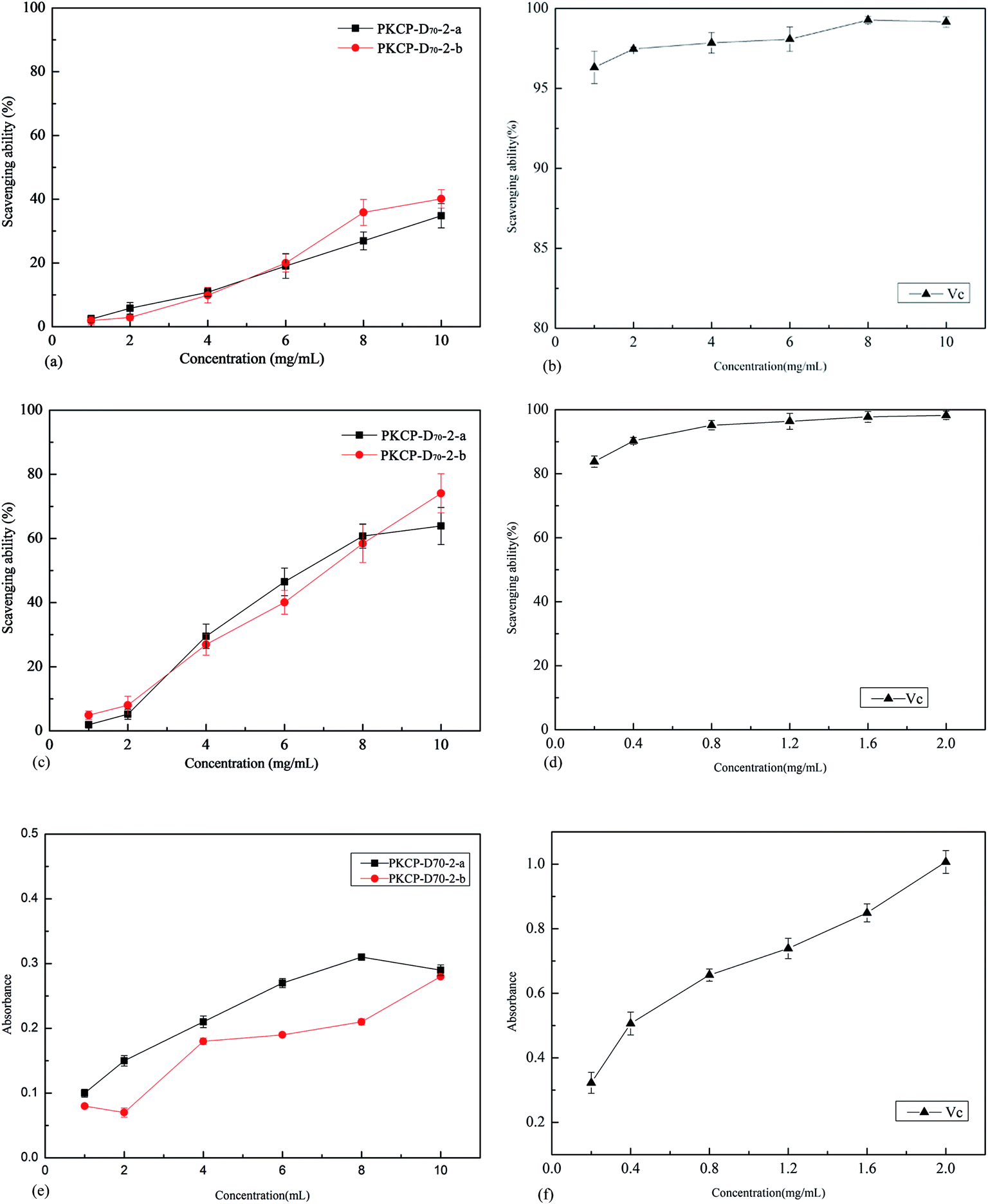 | ||
| Fig. 9 Antioxidant ability of PKCP-D70-2-a, PKP-D70-2-b and Vc, (a) ABTS˙+ radical scavenging ability of PKCP-D70-2-a and PKP-D70-2-b, (b) ABTS˙+ radical scavenging ability of Vc, (c) OH radical scavenging ability of PKCP-D70-2-a and PKCP-D70-2-b, (d) OH radical scavenging ability of Vc, (e) reducing power of PKCP-D70-2-a and PKP-D70-2-b, (f) reducing power of Vc. Values for the different samples at the same concentration with different lower case letters are significantly different at p ≤ 0.05. Values for the same sample at the different concentrations with different upper case letters are significantly different at p ≤ 0.05. Data are shown as means ± SD of triplicates (n = 3). | ||
The results of the ABTS+ radical scavenging assay are shown in Fig. 9a and b. The scavenging ability of the Vc for the ABTS+ radical was directly proportional to its concentration (1.0–10.0 mg L−1), while the IC50 value of the Vc is only just 0.01 mg mL−1. The ABTS+ radical scavenging ability of the PKCP-D70-2-a and the PKCP-D70-2-b were relatively low because their IC50 values were much higher than that of the Vc.
Many of the desirable properties have been correlated with structural features, such as molecular weight, type, and sequence distribution of sugar residues in the polysaccharide together with the presence (or the absence) of branching and the charged functional groups.57,58
The results of the hydroxyl radical scavenging assay are illustrated in Fig. 9c and d. The scavenging ability of the Vc for the hydroxyl radical was directly proportional to its concentration (0.2–2.0 mg L−1). Fig. 9c shows that the maximum hydroxyl radical scavenging ability of PKCP-D70-2-a and the PKCP-D70-2-b was approximately 63.9% and 74.1% at 2.0 mg L−1, and their IC50 were approximately 7.1 and 6.5 mg mL−1. In the low concentration (1.6–2.0 mg L−1), the scavenging ability of the Vc reached nearly 100%, and it's IC50 was just 0.25 mg L−1. Obviously, the hydroxyl radicals scavenging ability of PKCP-D70-2-a and the PKCP-D70-2-b was relatively low, compared with that of Vc. Moreover, the hydroxyl radical scavenging ability of PKCP-D70-2-b was stronger than that of the PKCP-D70-2-a. The results relate to the helical structure of polysaccharide; because the PKCP-D70-2-a and the PKCP-D70-2-b exhibited a triple helix structure whereas the PKCP-D70-2-b offered considerable helical configuration structures.
3.11. Evaluation of antioxidant ability
The phosphate group has valuable nucleophilic ability and an excellent capability to chelate metal ions, which indicate that polysaccharide with high phosphate group has a beneficial capability for scavenging hydroxyl free radicals.60 Previous research Wang et al. (2009) confirmed that the antioxidative ability of sulfated polysaccharide is more stronger.61 The content of phosphate group of the PKCP-D70-2-b was 0.051% higher than that of the PKCP-D70-2-a. However, the content of the sulfate group in the PKCP-D70-2-b was 0.19% lower than that of the PKCP-D70-2-a (Table 2). This outcome, when compared with the phosphate group, was due to sulfate groups not having robust chelating metal ion ability. Therefore, they have no obvious scavenging free radicals ability.The PKCP-D70-2-a molecular weight was 3048 kDa, and the relative molecular weight of the PKCP-D70-2-b barely attained 2548 kDa. Thus, the viscosity of the PKCP-D70-2-b, in an aqueous solution, was relatively small and more helpful to dissolve. As well, it displayed an enhanced scavenging hydroxyl and ABTS radical ability. The antioxidative ability of the PKCP-D70-2-b was slightly higher than that of the PKCP-D70-2-a, particularly under the concentration of 8–10 mg mL−1.
The influence of the glycosidic bond configuration on the oxidative capacity of polysaccharide largely, when compared to beta polysaccharide and polysaccharide with alpha configuration, has better biological activity.62 In this research, the PKCP-D70-2-b contained three kinds of β-glycosidic bonds and two kinds of α-configuration of the glycosidic bond. When compared to the PKCP-D70-2-b, the PKCP-D70-2-a contained a β-configuration of the glycosidic bond and two α-configurations of the glycosidic bond. Thus, by having a more meaningful β-configuration, the PKCP-D70-2-a gives it greater reducing power compared to the PKCP-D70-2-b.
The effect of glucose and mannose on the ability of the free scavenging radical was significant. It can be seen that the content of mannose and glucose in the PKCP-D70-2-b was higher than that of the PKCP-D70-2-a, the molar ratio was 15.9, 9.9. Consequently, this result confirmed that the PKCP-D70-2-b has effective antioxidant ability. This study is consistent with Rao et al.'s results.63
The branch degree of polysaccharide has a pronounced influence on its biological activity. Polysaccharide normally has a beneficial biological activity because of its appropriate branch degree. However, should the branch degree be too high or too low, such will limit its biological activity. The sugar content of the PKCP-D70-2-a and the PKCP-D70-2-b accounted for similar proportions with the respective branch degree of 55.49% and 53.80%. There was no significant difference in the scavenging free radicals ability between the PKCP-D70-2-b and the PKCP-D70-2-a. This may be attributed to the fact that branching has no significant influence on their antioxidant activities.
Polysaccharides with three helical conformations usually have strong biological activity. Both the PKCP-D70-2-a and the PKCP-D70-2-b demonstrated a triple helix conformation. Whereas the PKCP-D70-2-b had a larger conformation proportion of triple helix, there was a main free curl conformation in the PKCP-D70-2-a. Consequently, the free radicals scavenging ability of the PKCP-D70-2-b was stronger. Nevertheless, the PKCP-D70-2-a provides a stronger reducing power when compared to thePKCP-D70-2-b. Therefore, we conclude that all polysaccharides with a three helical conformation have a robust biological activity.
4. Conclusions
In this study, the PKCP-D70-2-a and the PKCP-D70-2-b were isolated from the Pinus koraiensis pinecone. The monosaccharide composition of the PKCP-D70-2-a was different from that of PKCP-D70-2-b. The existence of O-glycopeptide bond in PKCP-D70-2-a and PKCP-D70-2-b was demonstrated by β-elimination reaction. PKCP-D70-2-a and PKCP-D70-2-b were connected to the β-type glycosidic bond. The PKCP-D70-2-a and the PKCP-D70-2-b exhibited triple helix structures, whereas PKCP-D70-2-b presented considerable helical configuration structures.PKCP-D70-2-a and PKCP-D70-2-b offer both β-D-pyran-type and α-D-pyran-type sugar rings. PKCP-D70-2-a also shows a glycosidic bond 3,4)-α-Galp-(1→), →4)-α-D-Galp-(1→ and 4,6)-β-Glcp-(1→4), as the main link. Limited 1→3, 1→3,6, 1→2,3, 1→2,4, 1→3,4, 1→2,3,4 glycosidic galactose, pyranose, and arabinose bonds were connected. The PKCP-D70-2-b mainly demonstrated β-D-Glcp-(1→, β-D-Gal link α-D-Gal-6 sulfate, →4)-β-D-GalpA6Me-(→, →4)-α-D-Glc-(1→, α-D-Glc-(1→) connection with limited supplementary glycosidic bonds. Part of structural features of PKCP-D70-2-a and PKCP-D70-2-b is predicable; an outcome requires further investigation. In the antioxidant assays, the polysaccharide fractions showed effective scavenging activities on the ABTS and hydroxyl radicals, their antioxidant capabilities decreased in different order. Therefore, although the polysaccharide fractions had little effect on superoxide radical scavenging, this finding concerning the structure of the PKCP-D70-2-a and the PKCP-D70-2-b polysaccharides explained the reason why such polysaccharides may function with different antioxidant activities.
The implications and results from this research confirm that extract, isolation and purification polysaccharides from P. koraiensis pinecone is an efficient and effective way to make use of the P. koraiensis pinecone as a novel human medicinal product. The results also provide the first report on Pinus koraiensis pinecone polysaccharides and generate the need for further investigative research regarding the structure–activity relationship in the functional food industry.
Abbreviations
| PKCP | Pinus koraiensis cone polysaccharides |
| PKCP-D70 | The polysaccharides grades of PKCP by 70% ethanol |
| PKCP-D70-2-a and PKCP-D70-2-b | The polysaccharides isolated from the PKCP-D70 by the DEAE cellulose-52 and the Sephadex G-100 |
| Glc | Glucose |
| Gal | Galactose |
| Rha | Rhamnose |
| Ara | Arabinose |
| Man | Mannose |
| A | Uronic acid derivative |
| p | Pyranoside |
| f | Furanoside |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| TFA | Trifluoroacetic acid |
| ABTS | 2,2-Azino-bis(3-ethylbenzothiazoline-6-sulphonate) |
| TCA | Trichloroacetic acid |
| MW | Molecular weight |
| GPC | Gel-permeation chromatography |
| Tr | Retention time |
Acknowledgements
The National Natural Science Foundation of China (No. 31401483) and the Post-Doctoral Fund of Heilongjiang Province (LBH-Z14098) supported this work. The authors also are thankful for the Fundamental Research Funds for the Central Universities (Grant No. HIT. NSRIF. 2017025). The authors express their thanks to Professor Bob Tuck from Australia who edited and refined the paper.References
- J. H. Lee, H. Y. Yang, H. Sub Lee and S. K. J. Hong, J. Microbiol. Biotechnol., 2008, 18, 497–502 CAS.
- X. Yang, Y. Ding, Z. H. Sun and D. M. Zhang, Acta Pharmacol. Sin., 2005, 40, 435–437 CAS.
- K. Watanabe, F. Momose and H. Handa, Biochem. Biophys. Res. Commun., 1995, 214, 318–323 CrossRef CAS PubMed.
- P. Rohdewald, Int. J. Clin. Pharmacol. Ther., 2002, 40, 158–168 CrossRef CAS PubMed.
- G. H. Yen, P. D. Pin-Der Duh, D. W. Huang, C. L. Hsu and T. Fu Yu-Chi, Food Chem. Toxicol., 2008, 46, 175–185 CrossRef CAS PubMed.
- K. Li, T. Zhang, Z. Han, D. Gao and F. Zheng, Yakugaku Zasshi, 2007, 127, 1145–1151 CrossRef CAS.
- S. P. Potta, M. X. Doss, J. Hescheler and A. Sachinidis, Drug Des. Rev., 2005, 2(7), 85–91 CAS.
- N. Kadri, B. Khettal, A. Adjebli, T. Creseteil, R. Yahiaoui-Zaidi and V. Barragan-Montero, Ind. Crops Prod., 2014, 54, 6–12 CrossRef CAS.
- G. Asset, B. Staels, R. L. Wolff, E. Bauge, Z. Madj, J. C. Fruchart and J. Dallongeville, Lipids, 1999, 34, 39–44 CrossRef CAS PubMed.
- D. Bagchi, M. Bagchi, S. J. Stohs, D. K. Das, S. D. Ray, C. A. S. Kuszynski, S. Joshi and H. G. Pruess, Toxicology, 2000, 148, 187–197 CrossRef CAS PubMed.
- K. Li, Q. Li, J. Li, D. Gao, T. Zhang and Z. Han, Therapy, 2007, 4, 685–690 CrossRef CAS.
- J. H. Lee, S. M. Cho, K. S. Song, N. D. Hong and I. D. Yoo, Chem. Pharm. Bull., 1996, 44, 1093–1095 CrossRef CAS PubMed.
- A. Kilic, H. Hafizoglu, I. Tumen, I. E. Donmez, H. Sivrikaya, A. Sundberg and B. Holmbom, Wood Sci. Technol., 2010, 44, 523–529 CrossRef CAS.
- J. A. Micales, J. S. Han, J. L. Davis and R. A. Joung, Biodeterior. Res. 4, 1994, 4, 317–332 Search PubMed.
- R. B. Xu, X. Yang and J. Wang, Int. J. Biol. Macromol., 2012, 13, 14262–14277 CAS.
- Y. H. Li, W. W. Guan, N. Hao and H. Feng, Lishizhen Med. Mater. Med. Res., 2011, 22, 2127–2128 CAS.
- Y. Y. Li, X. Li, Y. J. Lian and B. Q. Li, Food Sci. Technol., 2012, 37, 210–213 CAS.
- M. Dubois, K. A. Gilles, J. K. Hamilton, P. A. Rebers and F. Smith, Anal. Biochem., 1956, 28, 350–356 CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- B. Nelly and A. H. Gustav, Anal. Biochem., 1973, 54, 484–489 CrossRef.
- P. Siddhuraju and K. Becker, J. Agric. Food Chem., 2003, 51, 2144–2155 CrossRef CAS PubMed.
- F. P. Qiu, L. Zhang and J. Yu, J. Changchun Univ. Technol., Nat. Sci. Ed., 2005, 26, 268–270 CAS.
- L. J. You, Q. Gao, M. Y. Feng, B. Yang, J. Y. Ren, L. J. Gu, C. Cui and M. M. Zhao, Food Chem., 2013, 138, 2242–2249 CrossRef CAS PubMed.
- F. Yuan, R. M. Yu, Y. Yin, J. R. Shen, Q. F. Dong, L. Zhong and L. Y. Song, Int. J. Biol. Macromol., 2010, 46, 436–439 CrossRef CAS PubMed.
- K. Das, Ind. Eng. Chem. Res., 2010, 49, 2176–2185 CrossRef CAS.
- Z. F. Wang, Y. Liu, Y. Sun, Q. Mou, B. Wang, Y. Zhang and L. J. Huang, Food Chem., 2014, 159, 137–142 CrossRef CAS PubMed.
- H. Zhang, Z. Y. Wang, L. Yang, X. Yang, X. Wang and Z. Zhang, Int. J. Mol. Sci., 2011, 84, 3202–3287 Search PubMed.
- Y. Yamamoto, T. Nunome, R. Yamauchi, K. Kato and Y. Sone, Carbohydr. Res., 1995, 275, 319–332 CrossRef CAS PubMed.
- C. G. Kumar, H. S. Joo, J. W. Choi, Y. M. Koo and C. S. Chang, Enzyme Microb. Technol., 2004, 34, 673–681 CrossRef CAS.
- S. Satitmanwiwat, K. Ratanakhanokchai, N. Laohakunjit, L. K. Chao, S. T. Chen, P. Pason, C. Tachaapaikoon and K. L. Kyu, J. Agric. Food Chem., 2012, 60, 5423–5430 CrossRef CAS PubMed.
- D. Rout, S. Mondal, I. Chakraborty and S. Islam, Carbohydr. Res., 2008, 343, 982–987 CrossRef CAS PubMed.
- F. Y. Meng, Y. L. Ning, J. Qi, H. Zhou, J. Jie, J. J. Lin, Y. J. Huang, F. S. Li and X. H. Li, Int. J. Mol. Sci., 2014, 15, 5140–5142 CrossRef PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- N. Smirnoff and Q. J. Cumbes, Phytochemistry, 1989, 28, 1057–1060 CrossRef CAS.
- J. F. Yuan, Z. Q. Zhang and Z. C. Fan, Carbohydr. Polym., 2008, 74, 822–827 CrossRef CAS.
- N. Pugh, S. A. Ross and H. N. ElSohly, Planta Med., 2001, 67, 737–742 CrossRef CAS PubMed.
- M. Sarkar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1976, 14, 919 Search PubMed.
- C. Hara, T. Kiho and S. Ukai, Carbohydr. Res., 1983, 117, 201–213 CrossRef CAS.
- J. P. Jiang, Z. Z. Yan and S. Z. Zhang, Acta Mycol. Sin., 1996, 15, 288–291 CAS.
- X. D. Li, J. Huazhong Agric. Univ., 2002, 31, 110–124 CAS.
- G. D. Manrique and F. M. Lajolo, Postharvest Biol. Technol., 2002, 25, 99–107 CrossRef CAS.
- L. R. Wen, Q. Gao, C. W. Ma, Y. Z. Ge, L. J. You, R. H. Liu, X. Fu and D. Liu, J. Funct. Foods, 2016, 20, 400–410 CrossRef CAS.
- C. Li, Q. Huang, X. Fu, X. J. Yue, R. H. Liu and L. J. You, Int. J. Biol. Macromol., 2015, 75, 298–305 CrossRef CAS PubMed.
- Z. Wang, F. Zhou and Y. Quan, Int. J. Biol. Macromol., 2014, 64, 139–143 CrossRef CAS PubMed.
- J. F. G. Vliegenthart, ACS Symp. Ser., 2006, 930, 1–19 CrossRef CAS.
- W. A. Bubb, Concepts Magn. Reson., Part A, 2003, 19, 1–19 CrossRef.
- M. Habibi and M. R. Mahrouz, Carbohydr. Polym., 2005, 60, 205–213 CrossRef.
- J. Velasco, H. Moll, Y. A. Knirel, V. Sinnwell, I. Moriyón and U. Zahringer, Carbohydr. Res., 1998, 306, 283–290 CrossRef CAS PubMed.
- B. H. Zhou, L. Yang, Y. Yuan, H. Shen, Q. Feng, Z. L. Guo and G. Liu, Clinical Hospital Pharmacological Journal, 2009, 29, 2002–2006 CAS.
- V. V. Golovchenko, R. G. Ovodova, A. S. Shashkov and Y. S. Ovodov, Phytochemistry, 2002, 60, 89–97 CrossRef CAS PubMed.
- P. K. Agrawal, Phytochemistry, 1992, 31, 330–3330 CrossRef.
- J. S. Maciel, L. S. Chaves and B. W. S. Souza, Carbohydr. Polym., 2008, 71, 559–565 CrossRef CAS.
- Y. Jing, X. Cui and Z. Chen, Carbohydr. Polym., 2014, 102, 288–296 CrossRef CAS PubMed.
- J. E. Li, S. W. Cui and S. P. Nie, Carbohydr. Polym., 2014, 105, 276–284 CrossRef CAS PubMed.
- J. Kumirska, J. Szafranek and M. Czerwick, Carbohydr. Res., 2007, 342, 2138–2143 CrossRef CAS PubMed.
- A. X. Luo, X. J. He, S. D. Zhou, Y. J. Fan, A. S. Luo and Z. Chun, Carbohydr. Polym., 2010, 79, 1014–1019 CrossRef CAS.
- M. Rinaudo, Food Hydrocolloids, 2001, 15, 433–440 CrossRef CAS.
- G. Muralikrishna and M. S. Rao, Crit. Rev. Food Sci. Nutr., 2007, 47, 599–610 CrossRef CAS PubMed.
- S. Meir, K. Kanner, B. Akiri and S. Philosoph-Hadas, J. Agric. Food Chem., 1995, 43, 1813–1819 CrossRef CAS.
- Z. S. Zhang, Chinese Academy of Sciences, 2010, 1, 20–24 Search PubMed.
- J. Wang, L. Liu, Q. B. Zhang, Z. S. Zhang, H. M. Qi and P. C. Li, Food Chem., 2009, 114, 1285–1290 CrossRef CAS.
- B. Mulloy, P. A. S. Mourao and E. J. Gray, J. Biotechnol., 2000, 77, 123–135 CrossRef CAS PubMed.
- R. S. P. Rao and G. Muralikrishna, Phytochemistry, 2006, 67, 91–99 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |