DOI:
10.1039/C6RA22719H
(Paper)
RSC Adv., 2016,
6, 103455-103462
Green synthesis of 1,4-benzodiazepines over La2O3 and La(OH)3 catalysts: possibility of Langmuir–Hinshelwood adsorption†
Received
11th September 2016
, Accepted 22nd October 2016
First published on 24th October 2016
Abstract
Lanthanum oxide [La2O3] and lanthanum hydroxide [La(OH)3] have been utilized as bases for the three-component reaction of aldehyde, dimedone and o-phenylenediamine for the synthesis of 1,4-benzodiazepine derivatives. The X-ray diffraction pattern shows pure phase formation of both catalysts. The basicity of La2O3 and La(OH)3 was measured by CO2-TPD (temperature programmed desorption) experiment. The measurements indicate the presence of basic sites that are useful for the reaction. Surface area measurements using Brunauer–Emmett–Teller (BET) reveal a low surface area for both the materials. TEM measurements indicate agglomerated particles with heterogeneous size distribution. The multicomponent reaction also proceeds smoothly in aqueous media as well as organic solvents. The heterogeneous catalyst was successfully recycled for 11 iterations without losing the catalytic activity. Several reactions were performed to understand the mechanism of the adsorption and the experiments suggest that the reaction may possibly proceeds by a Langmuir–Hinshelwood adsorption mechanism.
Introduction
Multi-component reactions (MCR) provide an attractive synthetic strategy to develop structurally diverse organic molecules.1,2 Simple experimental procedures and one-pot syntheses make MCRs highly sought after for the generation of several different structural variants of organic compounds.3,4 MCRs with heterogeneous catalysts offer great advantages due to atom economy, catalyst recyclability, low catalyst loading and high synthetic yields apart from the benefits that it may offer to the environment.5–7 Some multi-component one-pot reactions involving heterogeneous catalysts include the synthesis of 1,4-dihydropyridines,8,9 xanthenes,10,11 spirooxindole-pyrimidines,12 pyrroles,13 quinoxalines,14 pyridines,15 isoquinolones16 and diazepines.17–20 Benzodiazepines are heterocyclic compounds with pharmacological applications21,22 involving anti-tumor and anti-parasitic activities.23–25 Synthesis of these biologically relevant benzodiazepine derivatives was achieved through ionic liquid catalysis,26 solid phase synthesis,5,27 organometallic reagents28–30 and by other methods.31 Previous examples of heterogeneous catalysts utilized for the synthesis of substituted benzodiazepines include ZnS nanoparticles, chitosan supported Fe3O4 nanoparticulate materials,32 silver salts of silico-tungstic acid, Sn(HPO4)2·H2O, MoO3–SiO2 bifunctional catalysts, mixed-metal oxides33–39etc. Solid-base catalysts involving alkaline earth metal oxides, alkali doped materials, transition metal oxides and hydrotalciates were also widely utilized for organic reactions.40–43 But these catalysts are limited by their recyclability. La2O3 and La(OH)3 are highly basic and cost effective basic materials which make them suitable for many organic reactions. La2O3 is commonly used as a support oxide in gas solid reaction in catalytic convertors and methane activation.44–47 Recently we demonstrated the utility of lanthanum oxide and lanthanum hydroxide as recyclable catalysts for Michael addition and Hantzsch reactions.48 The economic benefit offered by synthetic ease of the catalysts, recyclability and the removal of tedious purification procedures is key to the development of heterogeneous catalytic materials. Extending our earlier work, herein, we report one-pot green synthesis of benzodiazepine derivatives using recyclable La2O3 and La(OH)3 as heterogeneous catalysts. Mechanistic possibility involving Langmuir–Hinshelwood adsorption mechanism is also discussed.
Experimental section
All the reagents used in the synthesis were purchased from Aldrich and SD fine chemicals and were without any further purification. La2O3 and La(OH)3 that were used in the synthesis were prepared by solution combustion method using urea as a fuel.48 Briefly, La(NO3)3·6H2O (5 g) and urea (1.734 g) were dissolved in distilled water (20 mL) followed by heating in a muffle furnace at 500 °C. Dehydration of the solution followed by combustion gave a white powder that consisted of the mixed phases of La(OH)3, La2O3 and some unknown phases. Calcination of this powder at 700 °C for 10 h led to the formation of pure La2O3 phase. La2O3 converts back to pure La(OH)3 if left open in the atmosphere for 24 h. Re-calcination under similar conditions was required to regain the La2O3 phase. The basic property of La2O3 and La(OH)3 is investigated by using Temperature Programmed Desorption (TPD) with CO2 gas. To determine the CO2 desorption ability of the catalyst, flame ionization detector (FID) is used. 50 mg of the catalyst was placed in a quartz tube of (6 mm OD, 4 mm ID and 25 cm length) and flushed with N2 gas from room temperature to 150 °C with a flow rate of 30 mL min−1. This step was necessary to remove the moisture from the sample. After reaching the final temperature (150 °C), it was allowed to cool down to room temperature. The flow of nitrogen gas was stopped and the sample was flushed with CO2 at the same flow rate for 45 min. To ensure removal of any residual gaseous phase or weakly adsorbed CO2 gas, the sample was again flushed in nitrogen gas for 30 min at room temperature. The sample was then heated in the presence of nitrogen gas with the same flow rate from room temperature to 600 °C at the heating rate of 10 °C min−1 to desorb the gas. Water formation, particularly from La(OH)3, was removed by using anhydrous CaCl2 tube connected in the gas line before it enters the detector. The peak area recorded for each catalyst gave an indication of the amount of CO2 adsorbed on the catalyst and gives a measure of basicity of the catalyst.
Structural characterization of the catalysts before and after the reaction was carried out by X-ray diffraction (XRD) with Bruker D8 Discover diffractometer using Cu Kα in the Bragg angle range of 10–80°. BET surface area were estimated using N2 sorption isotherms obtained at 77 K using Micrometrics ASAP 2020 instrument. Samples were degassed at 90 °C for 12 h with a flow of N2 gas. Transmission electron microscopy (TEM) for morphological characterization was carried out using Philips, Tecnai 20 microscope at 200 kV by dispersing the powdered catalyst in methanol and depositing it on a copper grid. NMR spectra of the synthesized samples were recorded using Bruker Avance (500 MHz) spectrophotometer in CDCl3 or DMSO (D6) as the solvent using tetramethylsilane as internal standard.
Results and discussion
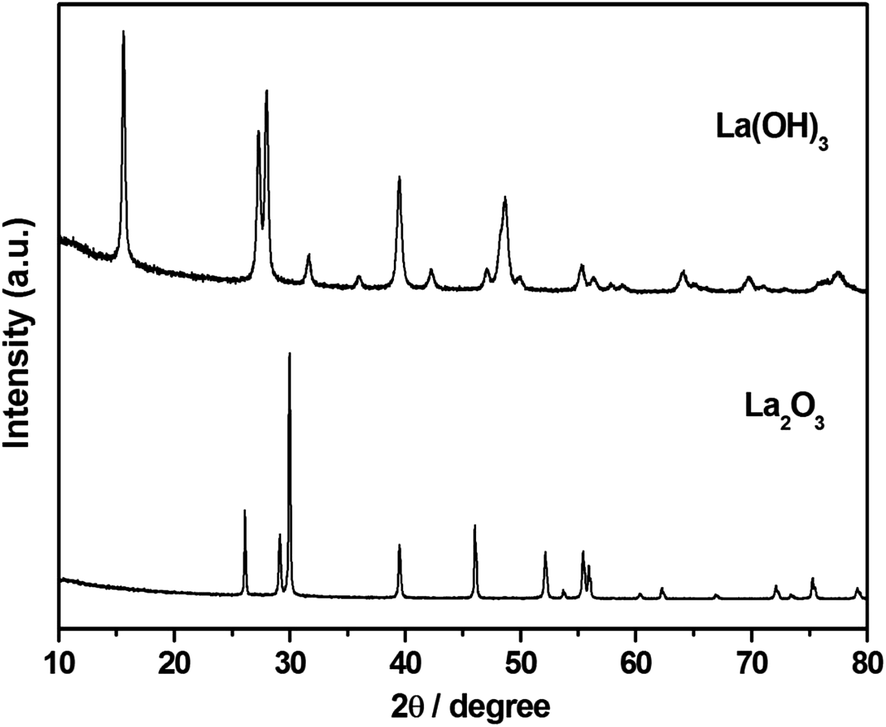
Structural characterization of La2O3 and La(OH)3 was carried out using XRD (Fig. 1). The XRD pattern shows sharp, intense peaks confirming high crystallinity for both La2O3 and La(OH)3. Pure phases of both La2O3 (JCPDS 73-2141) and La(OH)3 (JCPDS 13-1481) are obtained using the solution combustion method. The particle size was estimated by using the Scherrer equation,49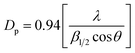 . The obtained particle size for La2O3 is 9.55 nm and 5.41 nm for La(OH)3. No changes in the crystal structure were noticed for La(OH)3 but La2O3 converts to La(OH)3 if kept open to atmosphere for about 24 h or after the organic synthetic reaction.48 The BET surface area obtained was 4.70 m2 g−1 and 11.0 m2 g−1 for La2O3 and La(OH)3 respectively. The requirement of synthesis of these compounds requires high temperature (700–800 °C) calcination for a long time (10 h) causing the surface area to reduce specifically for La2O3.
. The obtained particle size for La2O3 is 9.55 nm and 5.41 nm for La(OH)3. No changes in the crystal structure were noticed for La(OH)3 but La2O3 converts to La(OH)3 if kept open to atmosphere for about 24 h or after the organic synthetic reaction.48 The BET surface area obtained was 4.70 m2 g−1 and 11.0 m2 g−1 for La2O3 and La(OH)3 respectively. The requirement of synthesis of these compounds requires high temperature (700–800 °C) calcination for a long time (10 h) causing the surface area to reduce specifically for La2O3.
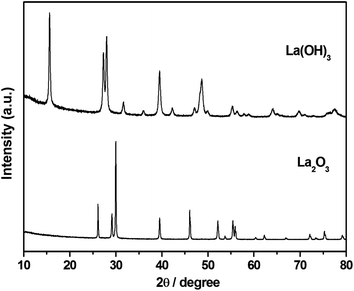 |
| | Fig. 1 XRD of combustion synthesized La2O3 and La(OH)3. La2O3 changes to La(OH)3 after the reaction while La(OH)3 retains the crystal structure. | |
Morphology of La2O3 and La(OH)3 (Fig. 2) was investigated using transmission electron microscopy (TEM). La(OH)3 shows agglomerates of highly heterogeneous particles. Boundaries of the particles are not visible throughout and shape also cannot be defined. Black spots possibly represent stacking of the particles over each other. TEM of La2O3 on the other hand shows particles with well-defined shapes growing in a particular manner. Particles are still agglomerated, yet distinguishable from each other. Although crystallinity cannot be established in the absence of lattice fringes, coupled with XRD data, we can confirm that both La(OH)3 and La2O3 are well crystalline in nature.
 |
| | Fig. 2 TEM of combustion synthesized La(OH)3 and La2O3 in the same magnification and resolution. Morphological differences are apparent. | |
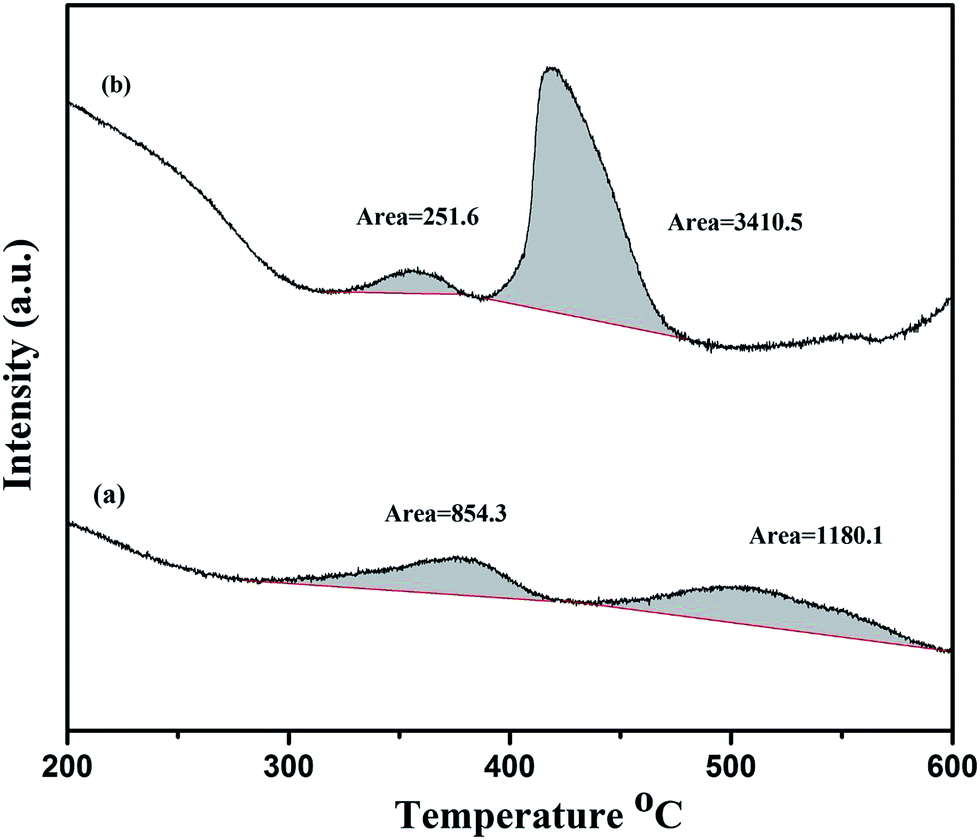
The CO2-TPD experiment is performed to estimate the basicity of La2O3 and La(OH)3 at room temperature. The obtained TPD curves are shown in Fig. 3. Since these compounds are basic in nature, they show considerable CO2 uptake even at room temperature. It has been previously reported that CO2 desorption temperature can classify the strengths of the basic sites.50 La2O3 shows two major peaks with peak temperatures 350 °C and 500 °C. This indicates that there are two different basic sites with varying strengths and the broad nature of the peak suggests that La2O3 has a wide range of distribution in basic site strength. Considering the amount of CO2 adsorbed over the surface, La(OH)3 appear more basic as the area under the peaks is almost two times higher than La2O3. However, La(OH)3 also has higher surface area (∼2.5 times) than La2O3 resulting more CO2 to adsorb on its surface. Further, considering the normalization of temperature and peak areas by BET surface area measurements, both the catalysts can be considered equally basic with only slight difference in the energetics at higher temperature.
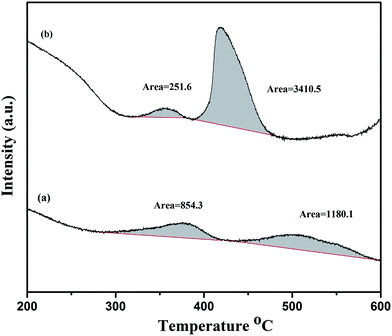 |
| | Fig. 3 Temperature programmed desorption of CO2. (a) La2O3 (b) La(OH)3. CO2 adsorption is carried out at room temperature followed by desorption in the temperature range of 200 to 600 °C. | |
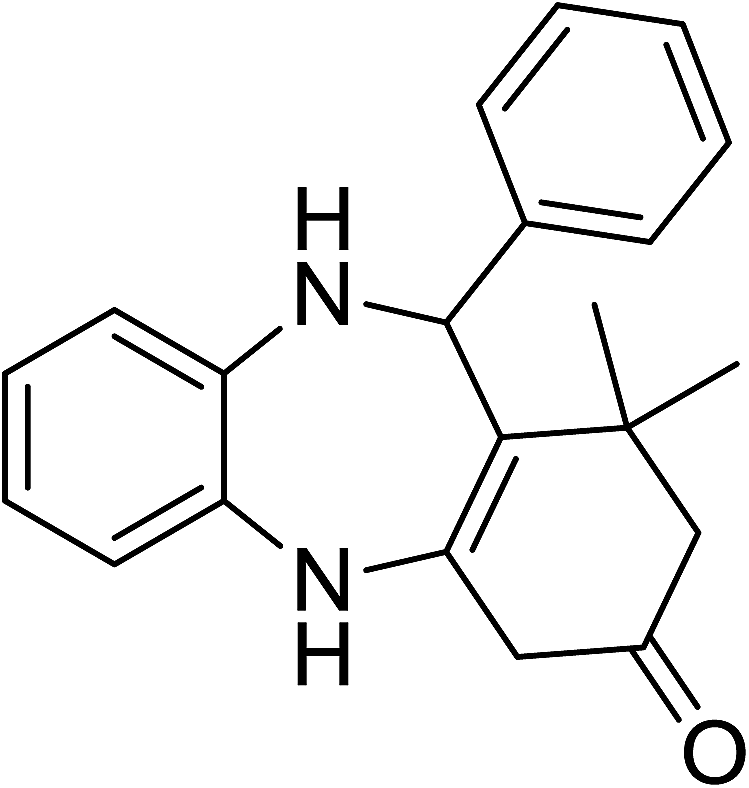
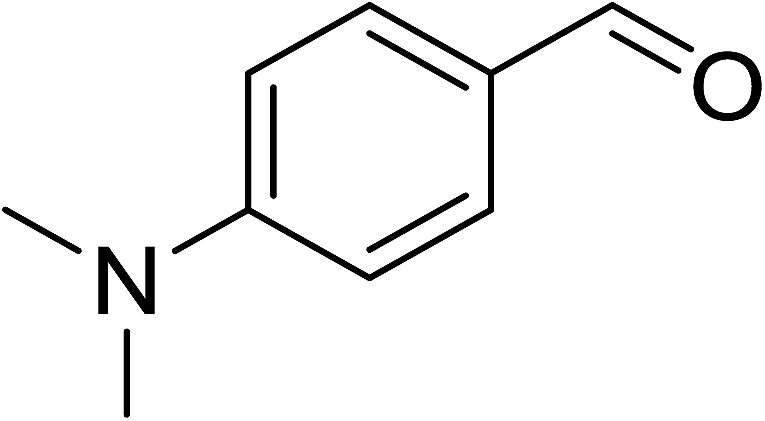
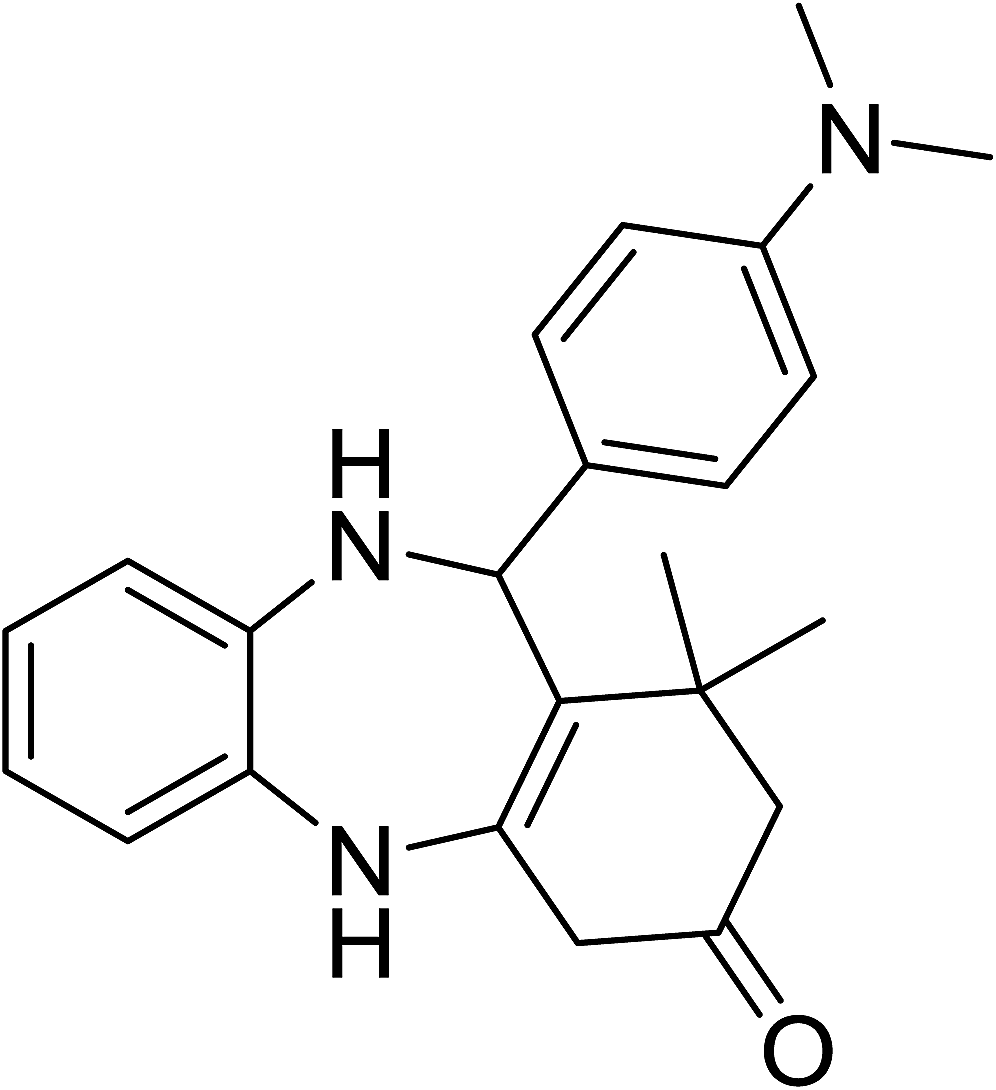
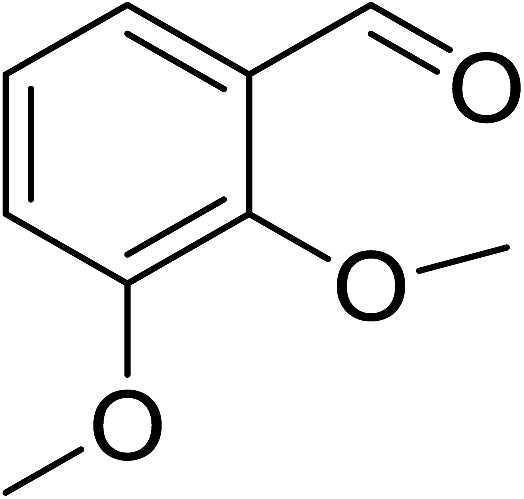
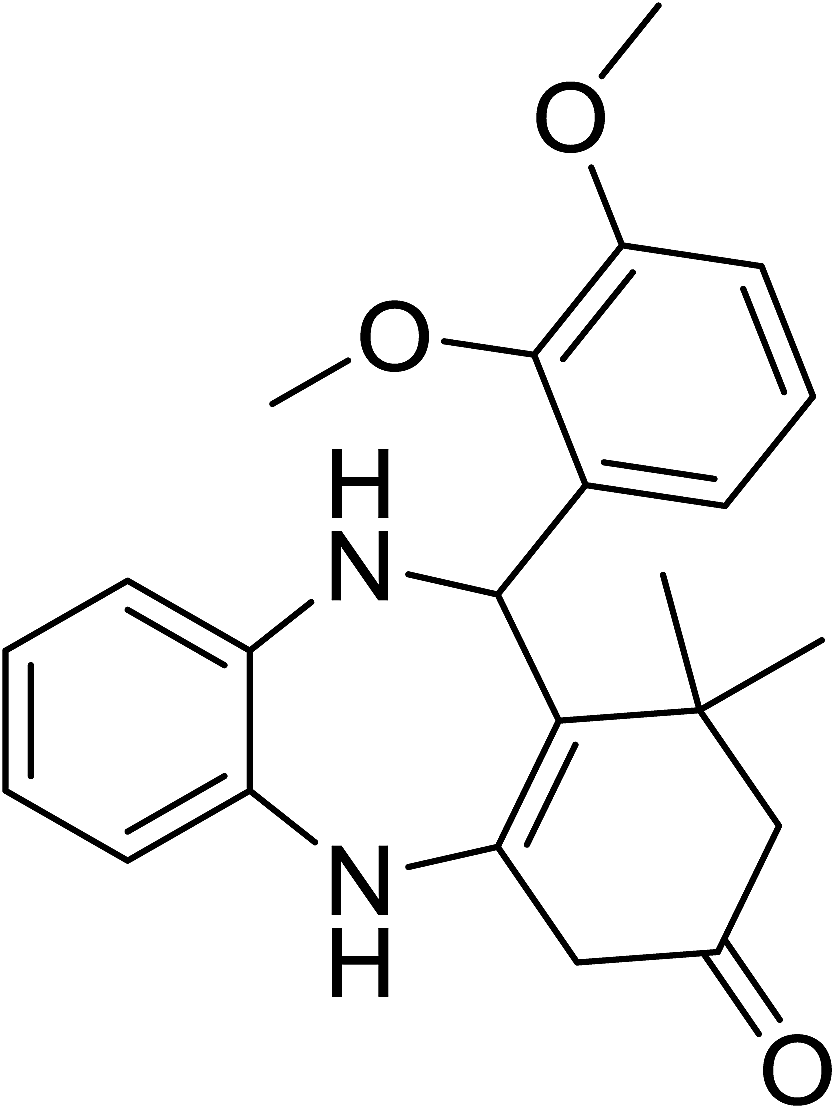
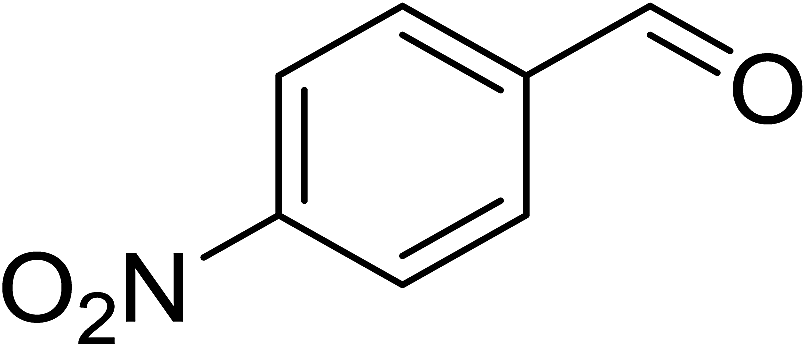
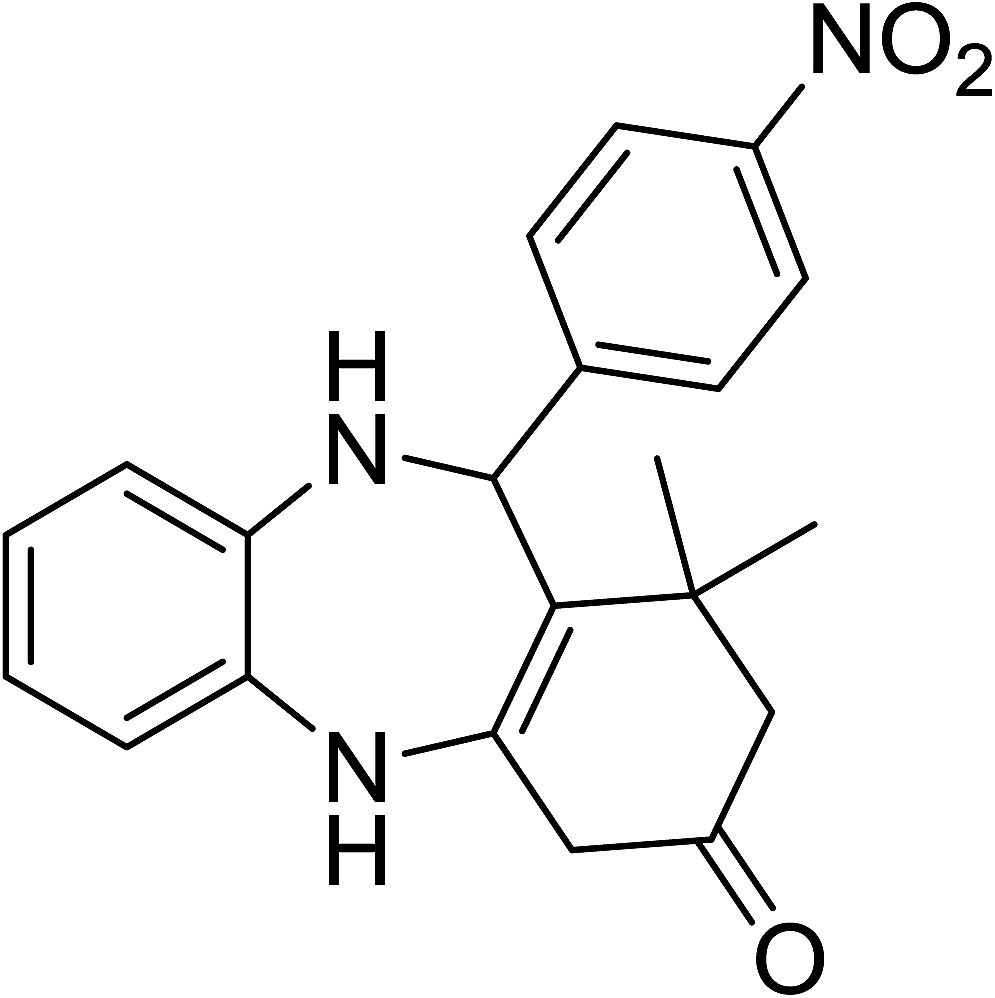
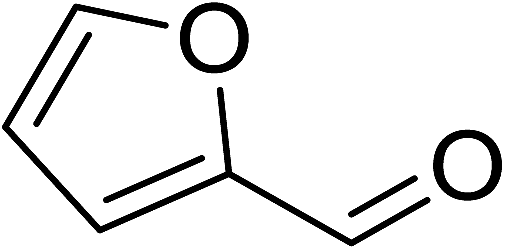
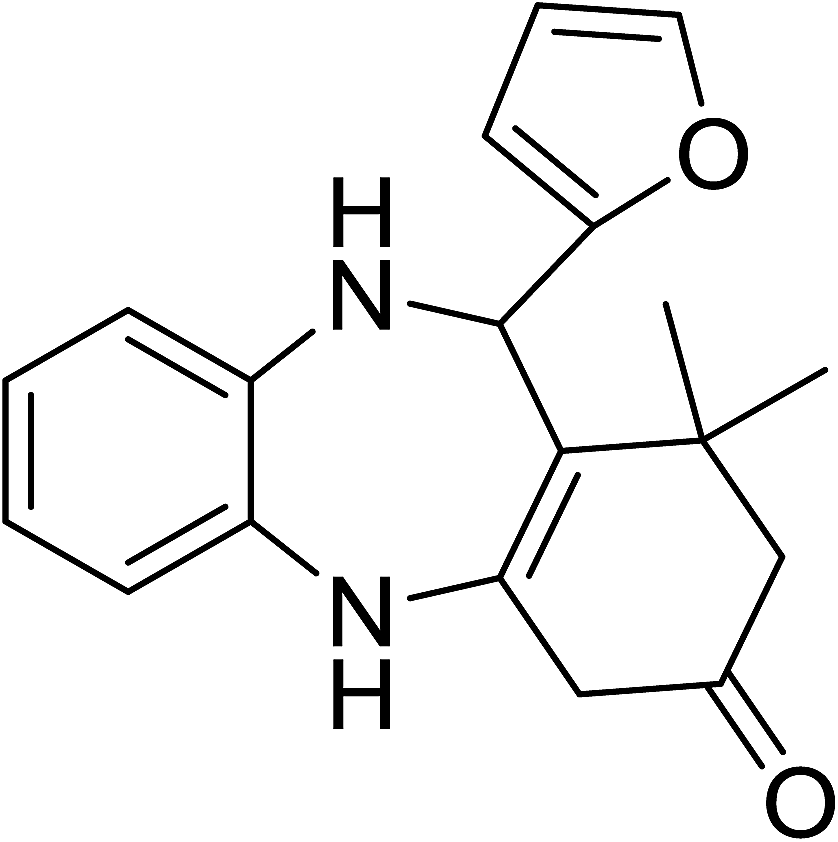
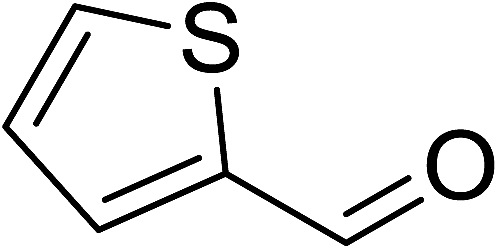
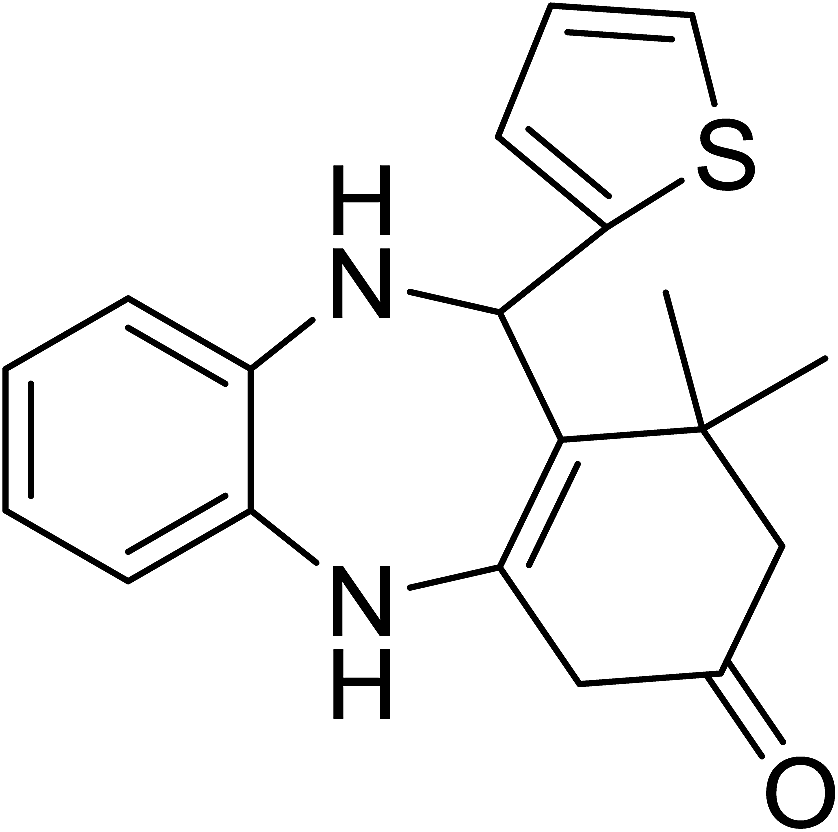
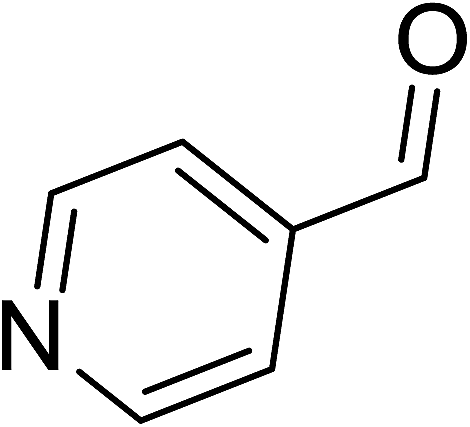
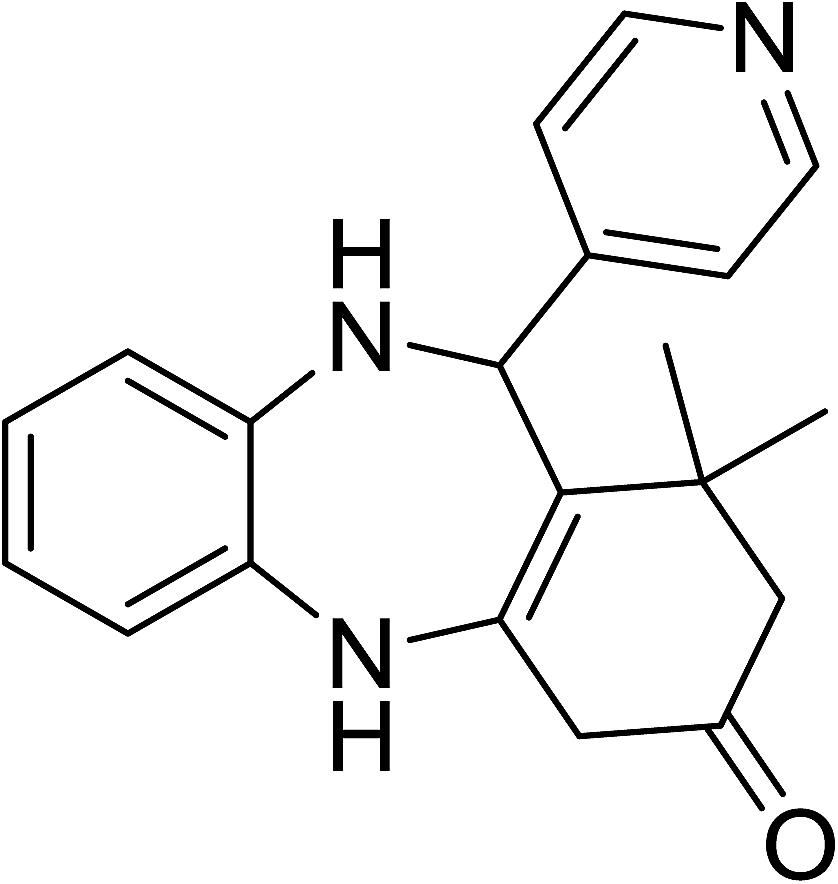
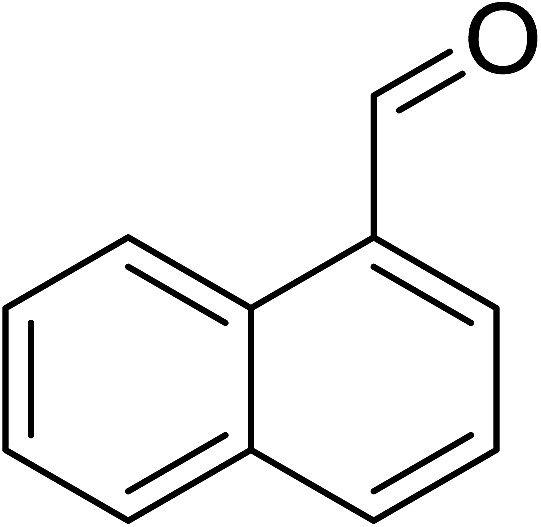
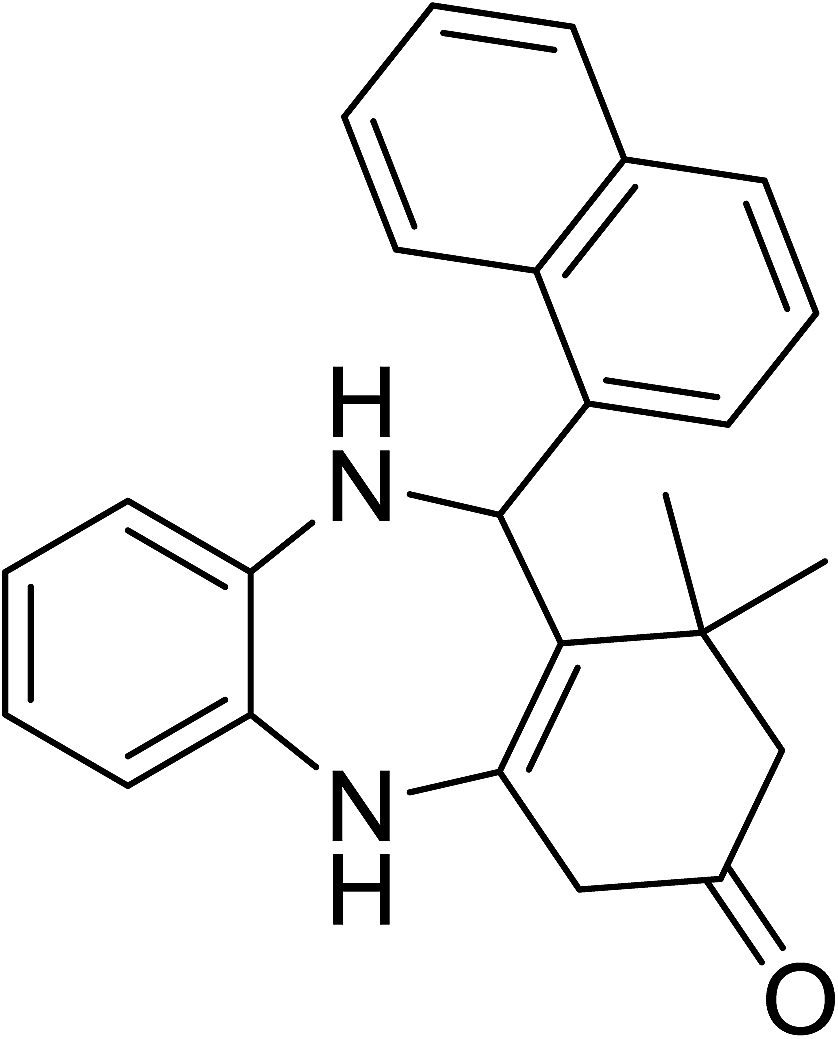
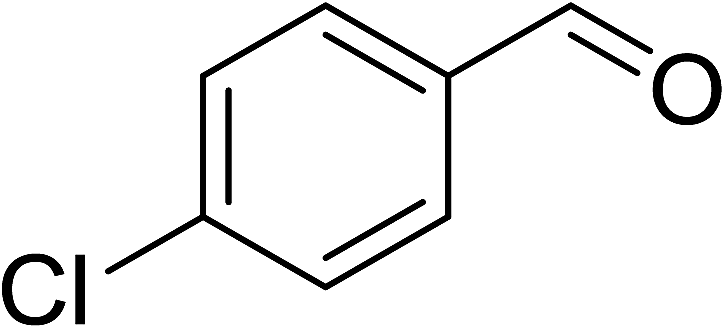
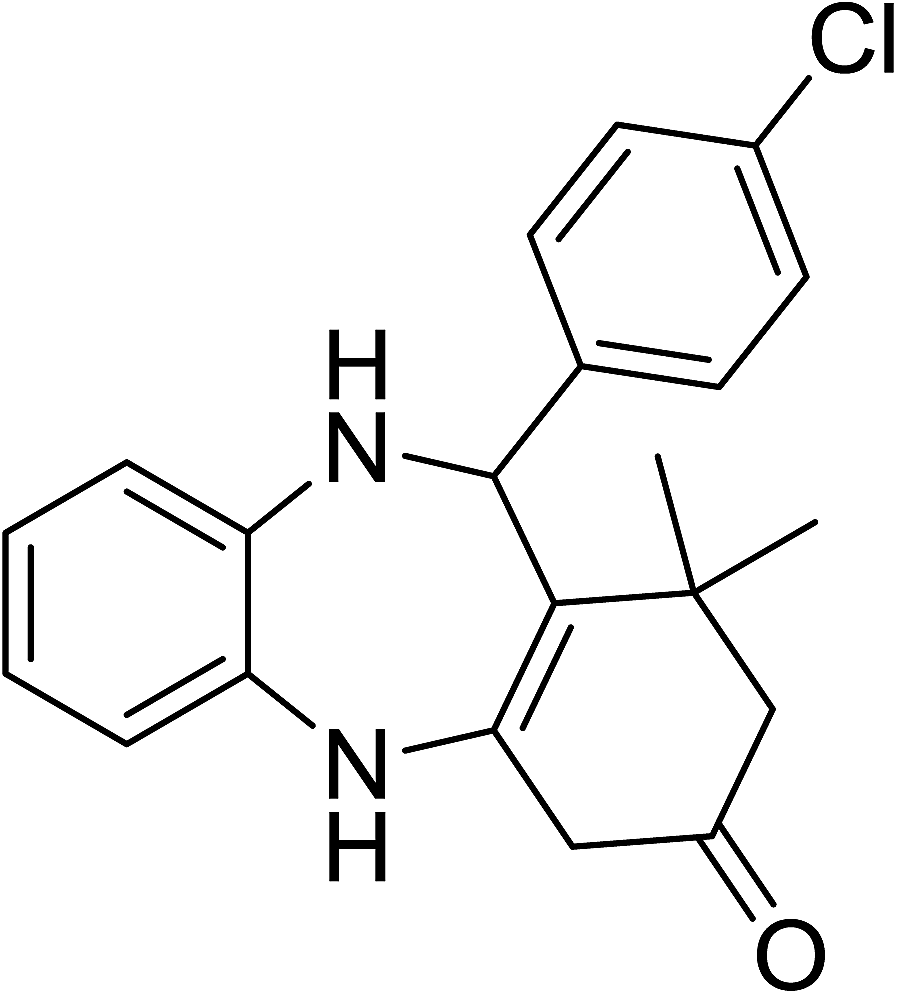
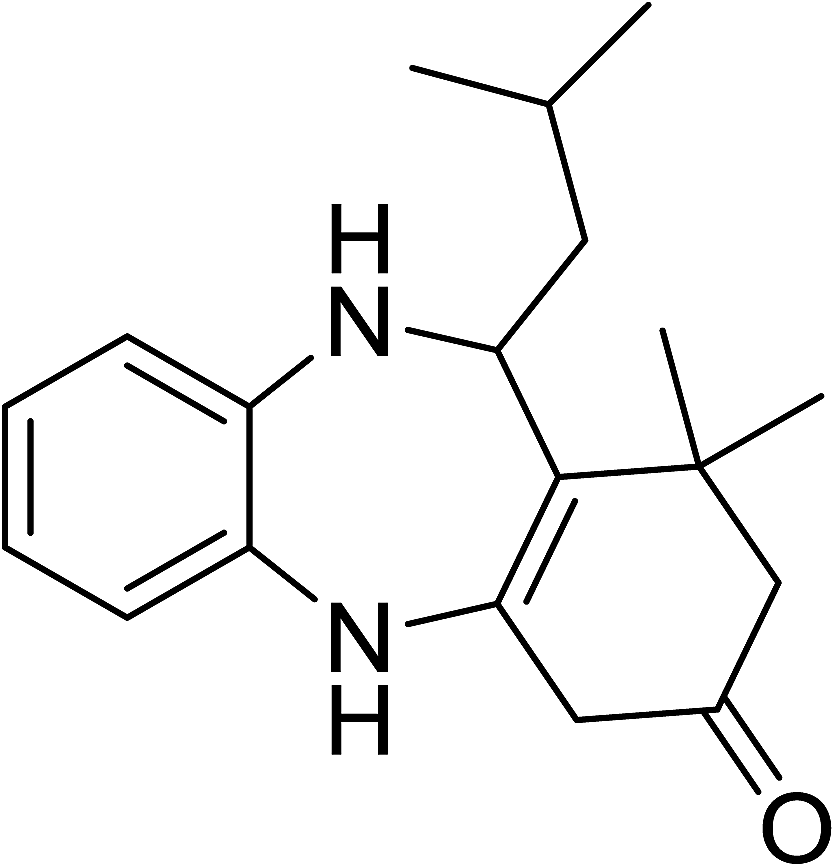
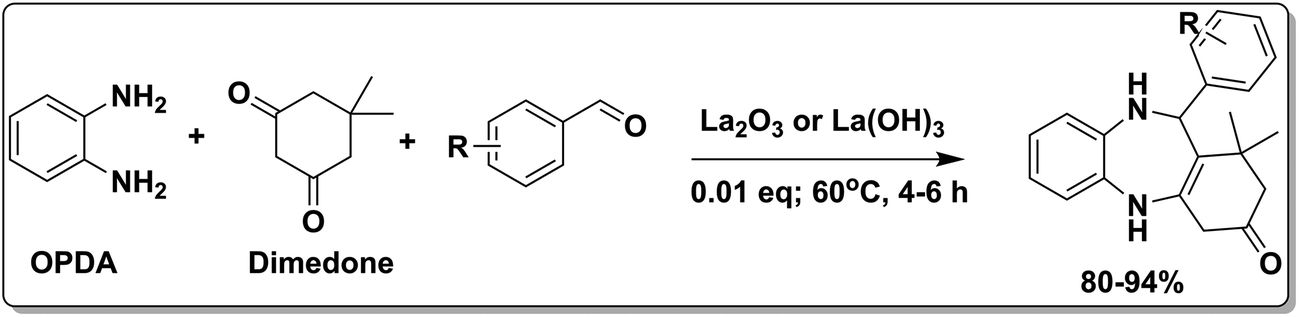
To demonstrate the utility of La2O3 and La(OH)3 as bases, we utilized one-pot synthesis of 1,4-benzodiazepines. Table 1 lists different reactants and the products obtained. In a typical procedure, 1 mmol of o-phenylenediamine (OPDA) was taken in a round bottom flask and appropriate equivalents of lanthanum oxide [La2O3] or lanthanum hydroxide [La(OH)3] in a suitable solvent. The contents were stirred for five minutes followed by the addition of 1 mmol of dimidone [Scheme 1]. The reaction mixture was heated to 60 °C for 15 minutes while being stirred until the observation of a new spot in thin layer chromatography [50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (ethyl acetate:petroleum ether)]. 1 mmol of a desired aldehyde derivative was then added slowly to the above reaction mixture. After completion of the reaction as monitored by TLC, the reaction mixture was cooled to room temperature followed by the addition of ice cold water. The contents were filtered using a Gooch crucible and the aqueous components were discarded. The organic components were extracted with ethyl acetate and dried using sodium sulfate. Concentration under reduced pressure followed by recrystallization in ethyl acetate yielded the desired product. No column chromatography was required. After structural confirmation of the product, 10 different experiments varying the aldehyde (aliphatic, heterocyclic or aromatic bearing electron donating or electron withdrawing groups) were performed (entries 1–10) as showed in the Table 1. In all cases, the reactions gave good to excellent yields revealing high activity of the catalysts under different substrate conditions. Similarity in the yield and reaction time confirms that the small difference in the basicity of La2O3 and La(OH)3 does not affect the catalytic property of both the materials and both the materials have similar type of useful basic sites. In general, it is expected that the basic sites of lanthanum oxide or lanthanum hydroxide catalyze coupling of dimedone with o-phenylenediamine and subsequently the formation of seven membered cyclic product.
:50 (ethyl acetate:petroleum ether)]. 1 mmol of a desired aldehyde derivative was then added slowly to the above reaction mixture. After completion of the reaction as monitored by TLC, the reaction mixture was cooled to room temperature followed by the addition of ice cold water. The contents were filtered using a Gooch crucible and the aqueous components were discarded. The organic components were extracted with ethyl acetate and dried using sodium sulfate. Concentration under reduced pressure followed by recrystallization in ethyl acetate yielded the desired product. No column chromatography was required. After structural confirmation of the product, 10 different experiments varying the aldehyde (aliphatic, heterocyclic or aromatic bearing electron donating or electron withdrawing groups) were performed (entries 1–10) as showed in the Table 1. In all cases, the reactions gave good to excellent yields revealing high activity of the catalysts under different substrate conditions. Similarity in the yield and reaction time confirms that the small difference in the basicity of La2O3 and La(OH)3 does not affect the catalytic property of both the materials and both the materials have similar type of useful basic sites. In general, it is expected that the basic sites of lanthanum oxide or lanthanum hydroxide catalyze coupling of dimedone with o-phenylenediamine and subsequently the formation of seven membered cyclic product.
Table 1 List of the reactions performed with the different aldehydes, their reaction times and the isolated product yield
| Entry |
Reactant A |
Reactant B |
Reactant C |
Product |
Time (h) La2O3/La(OH)3 |
Yield (%) La2O3/La(OH)3 |
| 1 |
OPDA |
dimedone |
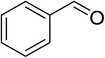
|
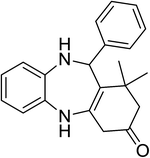
|
4/3.5 |
81/76 |
| 2 |
OPDA |
dimedone |
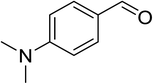
|
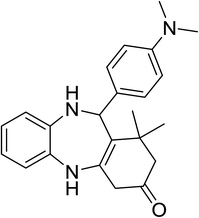
|
4.6/5 |
79/82 |
| 3 |
OPDA |
dimedone |
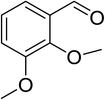
|
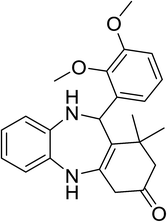
|
4.5/4.5 |
80/76 |
| 4 |
OPDA |
dimedone |
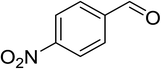
|
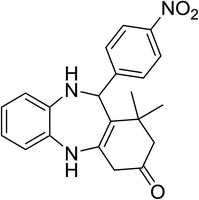
|
3.5/3 |
83/85 |
| 5 |
OPDA |
dimedone |
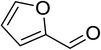
|
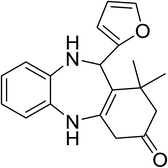
|
4.5/5 |
81/79 |
| 6 |
OPDA |
dimedone |
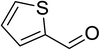
|
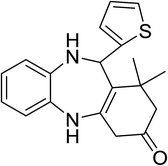
|
4.8/5 |
76/79 |
| 7 |
OPDA |
dimedone |
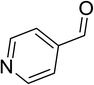
|
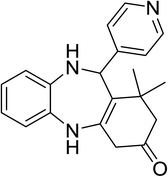
|
5/4.5 |
74/76 |
| 8 |
OPDA |
dimedone |
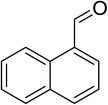
|
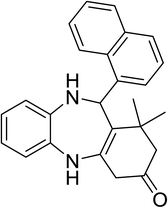
|
4/3.5 |
69/71 |
| 9 |
OPDA |
dimedone |
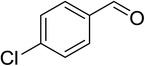
|
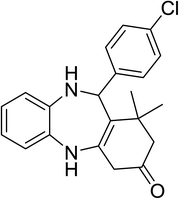
|
3.6/4 |
84/81 |
| 10 |
OPDA |
dimedone |
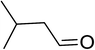
|
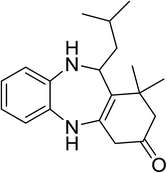
|
5/4.5 |
75/70 |
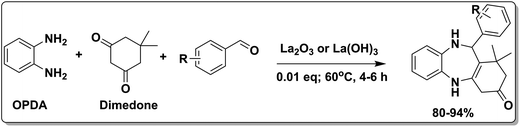 |
| | Scheme 1 Schematic of a synthetic strategy for preparation of 1,4-benzodiazepine derivatives. | |
Screening the solvent
In order to verify the medium effects on the reaction kinetics, the reactions were performed in different solvents as listed in Table 2. The solvent screening was done using benzaldehyde, dimedone and OPDA as the reagents (entry 1: Table 1) in the presence of 0.01 equivalents of catalyst [La2O3 or La(OH)3] at 60 °C. As shown in Table 2 poor yields were obtained with non-polar solvents (heptane, toluene, dioxane, THF). However, in the presence of polar solvents such as acetonitrile improved yields were obtained. Higher yields were obtained in the presence of DMSO, DMF and methanol with reduced reaction times. More importantly, the same reaction in aqueous media also gives excellent yields with comparable reaction kinetics demonstrating the greener approach of the catalysts utilized.
Table 2 Solvent screening in the presence of 0.01 equivalents of catalyst
| S. no. |
Solvent |
Reaction time (in hours) |
Yield (%) |
| 1 |
Heptane |
8.7 |
41 |
| 2 |
Toluene |
9 |
39 |
| 3 |
Dioxane |
7.8 |
45 |
| 4 |
THF |
8.3 |
48 |
| 5 |
Acetonitrile |
6 |
67 |
| 6 |
DMSO |
4.5 |
82 |
| 7 |
Methanol |
4 |
79 |
| 8 |
Water
|
4
|
81 |
Catalyst loading
The reaction of benzaldehyde, dimedone and OPDA at 60 °C with water was chosen to understand the optimum catalyst loading required for the reaction. Table 3 reveals success of the reactions with 0.001 equivalents of catalyst (entry 1), although the reaction times are on a higher side. Increase in the amount of catalyst (0.01 to 0.2 equivalents), results in the decrease of reaction times but product yields do not change much (Table 4).
Table 3 Effect of catalyst loading on reaction kinetics
| S. no. |
Catalyst loading |
Reaction time (hours) |
Product yield (%) |
| 1 |
0.001 |
8.5 |
79 |
| 2 |
0.005 |
7 |
75 |
| 3 |
0.01 |
4 |
81 |
| 4 |
0.05 |
3.5 |
80 |
| 5 |
0.1 |
3 |
78 |
| 6 |
0.2 |
3 |
82 |
Table 4 Mechanistic understanding of reactions
| Reaction no. |
Reagent addition sequence |
Reaction time (hours) |
Yield (%) |
| 1 |
A, B, C |
4 |
81 |
| 2 |
A, C, B |
5 |
56 |
| 3 |
B, A, C |
4.5 |
77 |
| 4 |
B, C, A |
— |
— |
| 5 |
C, A, B |
— |
— |
| 6 |
C, B, A |
— |
— |
Recyclability of the catalyst
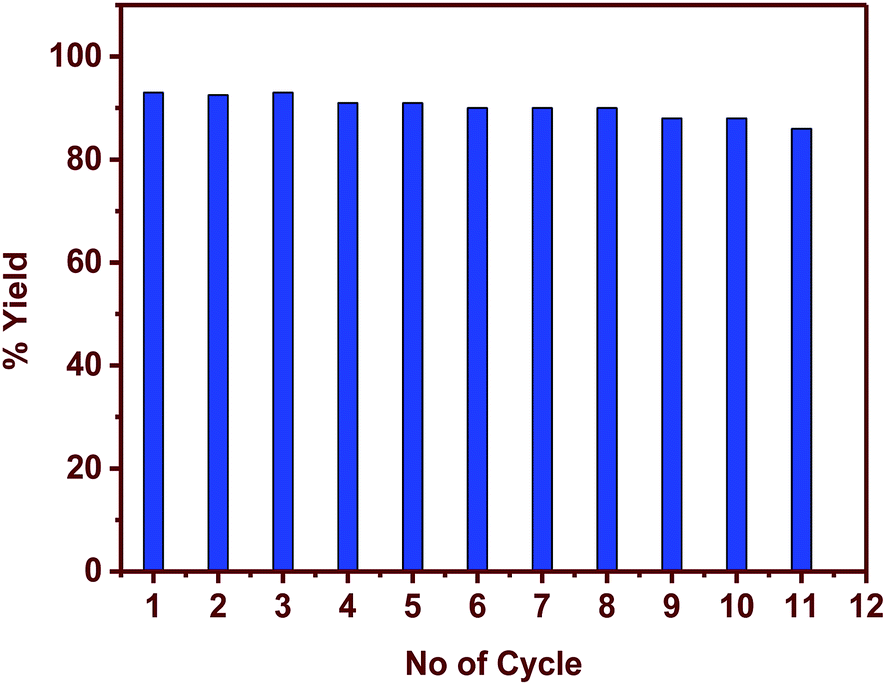
Compared to the homogeneous catalysts, heterogeneous catalysts have an advantage of reusability, hassle-free catalyst removal providing a cost-effective method in the industrial scale production. The reaction given in entry 1, Table 1 was used to understand the catalyst recyclability. Fig. 4 illustrates that the catalyst can be recycled for 11 cycles without any loss of activity. Further reactions could not be conducted because of the gradual loss of catalyst in each cycle.
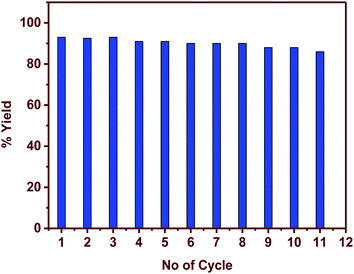 |
| | Fig. 4 Reusability of the catalyst and the corresponding yields (reaction conditions: temperature 60 °C, catalyst loading 0.01 equiv., solvent water, reaction entry 1, Table 1). | |
Understanding the adsorption of the reactants over the catalyst surface
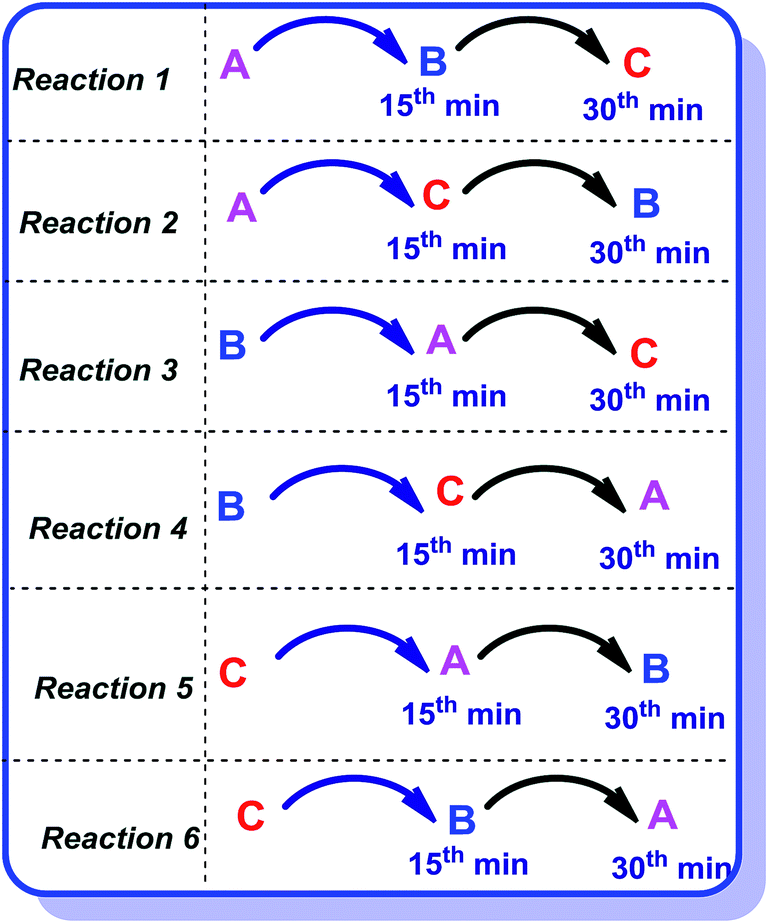
Heterogeneous catalysis is primarily based upon the adsorption phenomenon. There are two major mechanisms involved in the adsorption processes; Langmuir–Hinshelwood and Eley–Rideal mechanisms. In the former, all the three reactants are required to adsorb on the catalyst surface while in the latter, adsorption of all the reactants is not needed. To gain further insight of the type of adsorption mechanism, we performed one reaction (entry 1, Table 1) in six different ways [Fig. 5].
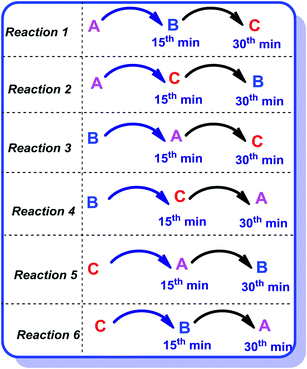 |
| | Fig. 5 Reactions performed to understand the mechanism involved. | |
The reactants OPDA, dimedone and benzaldehyde were labeled A, B and C respectively for simplicity. For this, water was used as the solvent with 0.01 equivalents of the catalyst and the reaction was performed at 60 °C. Each reactant was added step by step with a time gap of 15 minutes.
Method 1.
First A was added followed by addition of B and C with 15 minutes gap of each addition. We identified the disappearance of A in 15 minutes (by TLC), indicating the adsorption of A on the catalyst surface. After addition of B, in 15 minutes a new TLC spot was noticed that indicates the reaction has taken place between A and B, then we have added C. The reaction was smooth and completed in 4 h. The product was isolated, crystallized and characterized by NMR. The synthetic yield was 81%.
Method 2.
In this, the sequence of the reagents is A, C followed by B. A was added first and in 15 minutes C was added to the reaction mixture. After the addition of C, TLC showed three spots. Reagent B was subsequently added in 15 minutes and the product was isolated as described earlier in the experimental section. The overall reaction time is 5 h but the yields of this reaction dropped to 56%.
Method 3.
Sequence of reactants B, A and C were followed for Method 3, which gave similar reaction kinetics as reaction 1, but time of reaction was slightly extended to 4.5 h. However, the isolated product shows comparable yields (77%) similar to that obtained in Method 1.
Method 4.
In Method 4, the reagents were added in the sequence of B, C and A with 15 minutes gap for addition of each reagent. The crude reaction mixture gave a complex TLC and the product was not isolated as isolation was difficult.
Methods 5 and 6.
In 5th and 6th methods, C was the first reactant; both the reactions resulted in dark brown reaction mixture with traces of product formation. Many crystallization procedures were tried to isolate the product but were unsuccessful because of multiple product formation. From all these set of reactions, we concluded that the best order of reactant addition for smooth reaction is A followed by B and C. The details are summarized in Table 4.
Thus, we see that if we add A or B first (in any order), reaction occurs smoothly and reaction kinetics and the yield are similar. In all the other cases, either the reaction rate is affected or the reaction stops completely. Thus, adsorption of A and B is essential for the reaction to proceed. Initial adsorption of A and B leads to the formation of the intermediate and further addition of C yield the final reaction product. In fact adsorption of A seems most important as its adsorption in the beginning gives greater yield and faster kinetics (Method 1). It is important to note that if adsorption of only A and C was important and not B, reaction Method 2 would have given the same kinetics and yield as reaction 1, but it is significantly affected. If adsorption of only A and B was important and not C, reaction number 4, 5 and 6 would also give the yields and kinetics similar to reaction 1 but it does not even react. Finally, if adsorption of only B and C was important and not A, reaction number 6 will also give the reaction but it does not react either. Adsorption of C is further verified by carrying out a variation in Method 1. After adding A and B, the catalyst is removed from the reaction through simple filtration followed by the addition of C. After this, the reaction is allowed to continue in a usual manner as mentioned above. The time for the reaction was very large and the obtained product yield was also poor (15%). From these observations, we conclude that all the steps in the reaction require catalyst i.e., the adsorption of all the reactants was necessary. Therefore, the reaction mechanism may possibly be the Langmuir–Hinshelwood type where the adsorption of all the reactants is necessary. This is an important observation which makes this heterogeneous catalysis over La2O3/La(OH)3 different from homogeneous catalysis where the sequence of addition has a lesser role to play.
Conclusions
In conclusion, La2O3 and La(OH)3 are used as efficient catalysts for the synthesis of 1,4-benzodiazepine derivatives from various substituted o-phenylenediamines in water. This is a simple and efficient synthesis method for the three-component one-pot green synthesis using heterogeneous catalysis. Kinetics of the reactions is comparable with both La2O3 and La(OH)3 which means that the useful basic sites on the both the materials are similar. The reaction appears to follow the Langmuir–Hinshelwood adsorption model which is deciphered by running the reaction in different sequences. The use of water as a solvent enables greener synthetic methodology. Easy work up procedure, low catalyst loading, high product yields, avoidance of the column purification and catalyst recyclability provides an eco-friendly and environmentally benign process.
Acknowledgements
Authors thank DST (SR/S2/RJN-24/2012), (SB/S1/PC-81/2012) and IIT Gandhinagar for their financial support. Mr Vikas Patel from DST sponsored center SICART is gratefully acknowledged for TEM study.
References
- S. Brauch, S. S. Van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
-
J. Zhu and H. Bienaymé, Multicomponent reactions, John Wiley & Sons, 2006 Search PubMed.
- M. Jose Climent, A. Corma and S. Iborra, RSC Adv., 2012, 2, 16–58 RSC.
- A. Maleki, Z. Alrezvani and S. Maleki, Catal. Commun., 2015, 69, 29–33 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and L. L. Santos, Chem.–Eur. J., 2009, 15, 8834–8841 CrossRef CAS PubMed.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746–2762 RSC.
- M. Dixit, A. Menon, R. Baruah, A. Bhargav and S. Sharma, React. Kinet., Mech. Catal., 2015, 115, 611–624 CrossRef CAS.
- R. Pagadala, S. Maddila, V. D. B. C. Dasireddy and S. B. Jonnalagadda, Catal. Commun., 2014, 45, 148–152 CrossRef CAS.
- B. Karami, S. J. Hoseini, K. Eskandari, A. Ghasemi and H. Nasrabadi, Catal. Sci. Technol., 2012, 2, 331–338 CAS.
- T. S. Rivera, A. Sosa, G. P. Romanelli, M. N. Blanco and L. R. Pizzio, Appl. Catal., A, 2012, 443, 207–213 CrossRef.
- J. Deng, L.-P. Mo, F.-Y. Zhao, Z.-H. Zhang and S.-X. Liu, ACS Comb. Sci., 2012, 14, 335–341 CrossRef CAS PubMed.
- H. Ueda, M. Yamaguchi, H. Kameya, K. Sugimoto and H. Tokuyama, Org. Lett., 2014, 16, 4948–4951 CrossRef CAS PubMed.
- A. Huang, Y. Chen, Y. Zhou, W. Guo, X. Wu and C. Ma, Org. Lett., 2013, 15, 5480–5483 CrossRef CAS PubMed.
- A. Hakki, R. Dillert and D. W. Bahnemann, ACS Catal., 2013, 3, 565–572 CrossRef CAS.
- A. Maleki, RSC Adv., 2014, 4, 64169–64173 RSC.
- A. Maleki, Tetrahedron, 2012, 68, 7827–7833 CrossRef CAS.
- A. Maleki, Tetrahedron Lett., 2013, 54, 2055–2059 CrossRef CAS.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- C. S. Radatz, R. B. Silva, G. Perin, E. J. Lenardão, R. G. Jacob and D. Alves, Tetrahedron Lett., 2011, 52, 4132–4136 CrossRef CAS.
- W. Hunkeler, H. Mohler, L. Pieri, P. Polc, E. P. Bonetti, R. Cumin, R. Schaffner and W. Haefely, Nature, 1981, 290, 514–516 CrossRef CAS PubMed.
- W. Wisden and D. N. Stephens, Nature, 1999, 401, 751–752 CrossRef CAS PubMed.
- D. E. Thurston, H. Vassoler, P. J. M. Jackson, C. H. James and K. M. Rahman, Org. Biomol. Chem., 2015, 13, 4031–4040 CAS.
- N. Maheshwari, A. Goyal, S. Jain, D. Jain and N. S. H. N. Moorthy, Med. Chem. Res., 2014, 23, 765–774 CrossRef CAS.
- A. Kamal, R. V. Shetti, M. J. Ramaiah, P. Swapna, K. S. Reddy, A. Mallareddy, M. P. N. Rao, M. Chourasia, G. N. Sastry and A. Juvekar, MedChemComm, 2011, 2, 780–788 RSC.
- A. H. Jadhav, A. Chinnappan, R. H. Patil, S. V. Kostjuk and H. Kim, Chem. Eng. J., 2014, 240, 228–234 CrossRef CAS.
- B. Lemrová and M. Soural, ACS Comb. Sci., 2012, 14, 645–650 CrossRef PubMed.
- J. Qian, Y. Liu, J. Cui and Z. Xu, J. Org. Chem., 2012, 77, 4484–4490 CrossRef CAS PubMed.
- J. D. Neukom, A. S. Aquino and J. P. Wolfe, Org. Lett., 2011, 13, 2196–2199 CrossRef CAS PubMed.
- M. W. Gribble, J. A. Ellman and R. G. Bergman, Organometallics, 2008, 27, 2152–2155 CrossRef CAS PubMed.
- N. Kaur and D. Kishore, Synth. Commun., 2013, 44, 1375–1413 CrossRef.
- A. Maleki, N. Ghamari and M. Kamalzare, RSC Adv., 2014, 4, 9416–9423 RSC.
- M. Jeganathan and K. Pitchumani, ACS Sustainable Chem. Eng., 2014, 2, 1169–1176 CrossRef CAS.
- A. Maleki and M. Kamalzare, Catal. Commun., 2014, 53, 67–71 CrossRef CAS.
- A. Maleki and M. Kamalzare, Tetrahedron Lett., 2014, 55, 6931–6934 CrossRef CAS.
- H. Naeimi and H. Foroughi, New J. Chem., 2015, 39, 1228–1236 RSC.
- P. Hazarika, P. Gogoi, S. Hatibaruah and D. Konwar, Green Chem. Lett. Rev., 2011, 4, 327–339 CrossRef CAS.
- K. D. Parghi and R. V. Jayaram, Catal. Commun., 2010, 11, 1205–1210 CrossRef CAS.
- M. B. Gawande, P. S. Branco, K. Parghi, J. J. Shrikhande, R. K. Pandey, C. A. A. Ghumman, N. Bundaleski, O. M. N. D. Teodoro and R. V. Jayaram, Catal. Sci. Technol., 2011, 1, 1653–1664 CAS.
- A. Kawashima, K. Matsubara and K. Honda, Bioresour. Technol., 2008, 99, 3439–3443 CrossRef CAS PubMed.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- H. Hattori, Appl. Catal., A, 2001, 222, 247–259 CrossRef CAS.
- A. Guida, M. H. Lhouty, D. Tichit, F. Figueras and P. Geneste, Appl. Catal., A, 1997, 164, 251–264 CrossRef CAS.
- Y. Sekine, K. Tanaka, M. Matsukata and E. Kikuchi, Energy Fuels, 2009, 23, 613–616 CrossRef CAS.
- J. A. Kurzman, L. M. Misch and R. Seshadri, Dalton Trans., 2013, 42, 14653–14667 RSC.
- M. S. Palmer, M. Neurock and M. M. Olken, J. Am. Chem. Soc., 2002, 124, 8452–8461 CrossRef CAS PubMed.
- M. Dixit, A. Menon, R. Baruah, A. Bhargav and S. Sharma, React. Kinet., Mech. Catal., 2015, 115, 611–624 CrossRef CAS.
- B. P. Gangwar, V. Palakollu, A. Singh, S. Kanvah and S. Sharma, RSC Adv., 2014, 4, 55407–55416 RSC.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- O. V. Manoilova, S. G. Podkolzin, B. Tope, J. Lercher, E. E. Stangland, J.-M. Goupil and B. M. Weckhuysen, J. Phys. Chem. B, 2004, 108, 15770–15781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization spectra (1H-NMR, 13C-NMR and mass) of the synthesized molecules, catalyst removal procedure. See DOI: 10.1039/c6ra22719h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 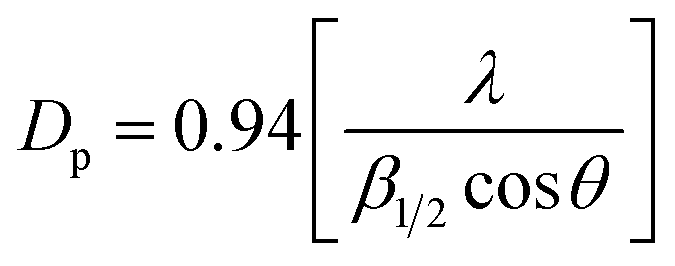 . The obtained particle size for La2O3 is 9.55 nm and 5.41 nm for La(OH)3. No changes in the crystal structure were noticed for La(OH)3 but La2O3 converts to La(OH)3 if kept open to atmosphere for about 24 h or after the organic synthetic reaction.48 The BET surface area obtained was 4.70 m2 g−1 and 11.0 m2 g−1 for La2O3 and La(OH)3 respectively. The requirement of synthesis of these compounds requires high temperature (700–800 °C) calcination for a long time (10 h) causing the surface area to reduce specifically for La2O3.
. The obtained particle size for La2O3 is 9.55 nm and 5.41 nm for La(OH)3. No changes in the crystal structure were noticed for La(OH)3 but La2O3 converts to La(OH)3 if kept open to atmosphere for about 24 h or after the organic synthetic reaction.48 The BET surface area obtained was 4.70 m2 g−1 and 11.0 m2 g−1 for La2O3 and La(OH)3 respectively. The requirement of synthesis of these compounds requires high temperature (700–800 °C) calcination for a long time (10 h) causing the surface area to reduce specifically for La2O3.