
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas-phase kinetic and mechanistic investigation of the OH radical and Cl atom oxidation of tetraethoxysilane
Ian
Barnes
* and
Peter
Wiesen
University of Wuppertal, School of Mathematics and Natural Sciences, Institute for Atmospheric and Environmental Research, Gauss Strasse 20, 42119 Wuppertal, Germany. E-mail: barnes@uni-wuppertal.de; Fax: +49 202 4392505; Tel: +49 202 439 2510
First published on 17th October 2016
Abstract
Rate coefficients have been determined in a large photoreactor for the reaction of OH radicals and Cl atoms with tetraethoxysilane (TEOS, (CH3CH2O)4Si) using a relative kinetic method and in situ FTIR spectroscopy for the analysis. Values of (2.99 ± 0.37) × 10−11 and (2.41 ± 0.47) × 10−10 cm3 per molecule per s were obtained for the reactions of OH and Cl with TEOS, respectively, at 298 K and atmospheric pressure. The products originating from the OH-radical and Cl-atom initiated oxidation of TEOS have also been investigated. Acetic acid (CH3C(O)OH) is observed as a major product in both cases with a molar yield of (95 ± 4)%. An oxidation mechanism is proposed to explain its formation and the possible identity of the co-product(s) is discussed.
Introduction
Silicon esters are silicon compounds that contain an oxygen atom located between the silicon atom and an organic group, i.e., Si–O-R, the most common of which is tetraethoxysilane (TEOS, (CH3CH2O)4Si). TEOS has many industrial applications in which the reactivity of the Si–OR bond is utilized whereby they can be roughly divided between whether the Si–OR bond is hydrolyzed or remains intact.1 A major use of TEOS is as a crosslinking agent in silicon polymers. It is employed in large quantities as a silicon source for the deposition of doped and undoped silicon dioxide films in the semiconductor industry and also as the silica source for the synthesis of zeolites.1 Other applications include its use in the production of aerogels and coatings for carpets and other objects.2Applications in which the Si–OR bond is hydrolyzed include the use as binders for foundry-mold sands used in investment and thin-shell castings; as binders for refractories; and as resins, coatings, sol–gel glasses, cross-linking agents, and adhesion promoters. Applications in which the Si–OR bond remains intact include the use as heat-transfer and hydraulic fluids.1
The production and many uses of TEOS can result in its release to the environment, in particular, through use as a chemical sol–gel, and in casting and glass frosting processes. To date, there do not appear to be any reports of atmospheric concentration measurements of TEOS in the literature.
Since TEOS has a vapour pressure of around 1.88 mm Hg at 25 °C (ref. 3) it is expected to exist mainly in the gas phase in the ambient atmosphere. This assumption is supported by gas/particle partitioning models of organic compounds.4 Evaluations of the environmental fate of TEOS suggest that any atmospheric emissions of TEOS will be mainly degraded by reaction with hydroxyl radicals.5 Using the structure activity relationship (SAR) estimation method AOPWIN for OH rate coefficients in the EPA Suite™,6 which is based on the SAR of Kwok and Atkinson,7 gives a value of 2.47 × 10−11 cm3 per molecule per s for the reaction of OH with TEOS at 298 K and atmospheric pressure. However, to the best of our knowledge, the rate coefficient for the gas-phase reaction of OH radicals with TEOS has never been determined experimentally and no studies exist in the literature on the atmospheric OH-radical initiated gas-phase degradation products.
Traditionally it has been thought that Cl atoms can only attain levels to make it a significant VOC oxidant in coastal and marine regions.8–11 However, in recent years evidence has been presented for Cl-atom mediated chemistry in continental regions remote from coastal and marine regions mainly involving the photolysis of nitryl chlorine as an early morning Cl-atom precursor.12–16 In addition, very recently high levels of molecular chlorine have been observed in the Arctic atmosphere.17
Reported here is a room temperature relative kinetic determination of the gas-phase rate coefficients for the reactions of OH radicals and Cl atoms with TEOS. In addition, an in situ FTIR semi-qualitative analysis of the reaction products produced in the OH-radical and Cl-atom mediated oxidation of TEOS is presented and discussed.
Experimental
The experiments were performed in a 1080 L quartz-glass photoreactor in synthetic air at a total pressure of 760 Torr (760 Torr = 101.325 kPa). The photoreactor has been described in detail elsewhere,18 thus only a brief description of the important elements of the reactor is given here. The reactor can be evacuated to 10−3 Torr using a pumping system consisting of a turbo-molecular pump backed by a double stage rotary fore pump. Homogeneous mixing of the reactants in the reactor is ensured by magnetically coupled Teflon mixing fans located inside the chamber. Two types of lamps have been used to photo-dissociate the radical/atom precursors: 32 super actinic fluorescent lamps (Philips TL 05/40 W: 320 < λ < 480 nm, λmax = 360 nm) and 32 low-pressure mercury lamps (Philips TUV/40 W, λmax = 254 nm). The lamps are wired in parallel and can thus be switched individually. They are distributed evenly around the photoreactor to ensure homogeneous irradiation within the reactor. A white type multiple-reflection mirror system set at a total optical path length of 484.7 ± 0.8 m is mounted inside the photoreactor. This optical system was used for sensitive monitoring of reactants and products in the IR spectral range 4000–700 cm−1. A Nicolet Nexus FT-IR spectrometer equipped with a KBr beam splitter and a liquid nitrogen cooled mercury–cadmium–telluride (MCT) detector was used to record IR spectra with a spectral resolution of 1 cm−1.The relative kinetic technique has been used to determine rate coefficients for the reactions of OH radicals and Cl atoms with tetraethoxysilane. Propene and 2-methylpropene have been used as reference compounds for the OH kinetic experiments whereas ethene and propene have been used for the Cl kinetic experiments. A number of factors have been considered in the choice of the reference compounds (i) that their rate coefficients are distinctly different from one another, (ii) that their rate coefficients are not more than a factor of five higher or lower than that of TEOS in order to minimize errors in the IR spectral analysis and (iii) their IR absorption bands overlap as little as possible with those of TEOS.
The photolysis of hydrogen peroxide using the mercury lamps has been used to produce OH radicals:
| H2O2 + hν (λ = 254 nm) → 2OH | (1) |
The photolysis of molecular Cl2 with the fluorescent lamps was used to generate Cl atoms:
| Cl2 + hν (320 < λ < 480 nm) → 2Cl | (2) |
The photolytically produced OH radicals or Cl atoms initiate the decay of TEOS and the reference compounds through the following reactions:
| OH/Cl + TEOS → products, kTEOS | (3) |
| OH/Cl + reference → products, kreference | (4) |
If the reference compound and the reactant are lost solely by reactions (3) and (4), then it can be shown that:
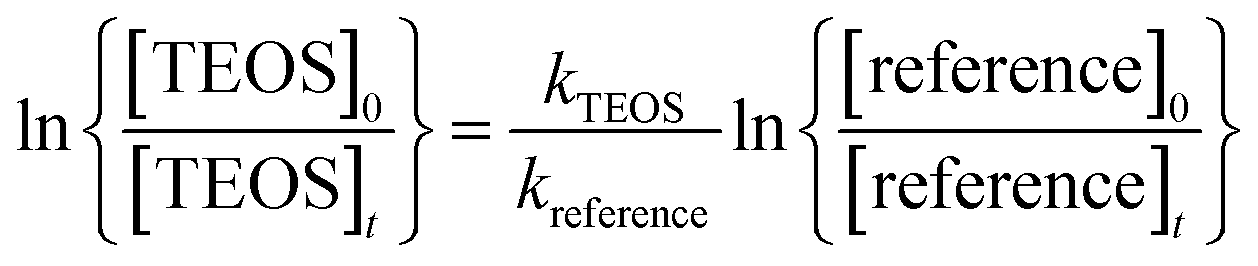 | (I) |
In order to verify TEOS is removed solely by reaction with either OH radicals or Cl atoms various tests were performed. Mixtures of TEOS and the reference compounds with either H2O2 or molecular chlorine were prepared and allowed to stand in the dark for 30 minutes which is the duration of a typical experiment. Loss of TEOS and the reference compounds over this time period was negligible showing that both wall loss and also reaction of the substances with the radical precursors is insignificant. Possible photolysis loss of TEOS and the reference compounds was tested by irradiating mixtures of TEOS and the reference compounds in air, in the absence of the radical precursors, with the fluorescent and mercury lamps. Neither type of lamp caused photolytic loss of TEOS or the reference compounds.
Calibrated reference spectra contained in the IR spectral databases of the laboratory in Wuppertal were used to follow the concentration–time behaviours of the reactants in the kinetic experiments. The spectral subtraction option in the OMNIC Software Suite 8.0 from Thermo Scientific was used for the spectral subtraction. The following infrared main absorption frequencies (in cm−1) were used to follow the concentration–time behaviours of the reactants: TEOS 2896 and 1116, ethene 950, propene 992 and 2-methylpropene 3087. Acetic acid the main identified reaction product was monitored and quantified using the absorption bands at 3581 and 1796 cm−1.
The initial concentrations of both TEOS and the reference compounds ethene, propene and 2-methylpropene were 2–4 ppmV (1 ppmV = 2.46 × 1013 molecule per cm3 at 298 K and 760 Torr of total pressure). The initial concentrations of H2O2 and Cl2 were typically around 10 and 5 ppmV, respectively. The experiments were performed in 760 Torr of synthetic air at (298 ± 2) K. In a typical experiment 60 interferograms were co-added per spectrum over a period of approximately 1 minute and 15–20 such spectra were recorded per experiment. The first 5 spectra were always recorded without lamps to check that wall loss of the reactants remained negligible.
The chemicals used in the experiments had the following purities as given by the manufacturer and were used as supplied: synthetic air (Air Liquide, 99.999%), Cl2 (Messer Griesheim, 2.8), ethene (Messer Schweiz AG, 3.5), propene (Air Liquide, 3.5), 2-methylpropene (Messer Griesheim, 99.99%), hydrogen peroxide (Interox, 85%), tetraethoxysilane (Sigma-Aldrich, 99.999%).
Results and discussion
Kinetic study
Fig. 1 shows exemplary plots of the kinetic data obtained from investigations on the reactions of OH radicals and Cl atoms with TEOS at (298 ± 2) K and atmospheric pressure for each of the reference compounds used in the respective investigations. For both reactions with each reference compound reasonable linear relationships with near zero intercepts were obtained in all experiments. Table 1 lists the rate coefficient ratios, kTEOS/kreference, obtained from least squares regression analysis of the plots of the kinetic data for all the experiments performed in the reactions of OH and Cl with TEOS. The errors are the 2σ standard deviations from linear regression analyses of the kinetic data plots. Also given in Table 1 are the average values of the rate coefficients for the reactions of OH and Cl with TEOS obtained from the individual measurements. The error given here encompasses the range of error given for the individual measurements.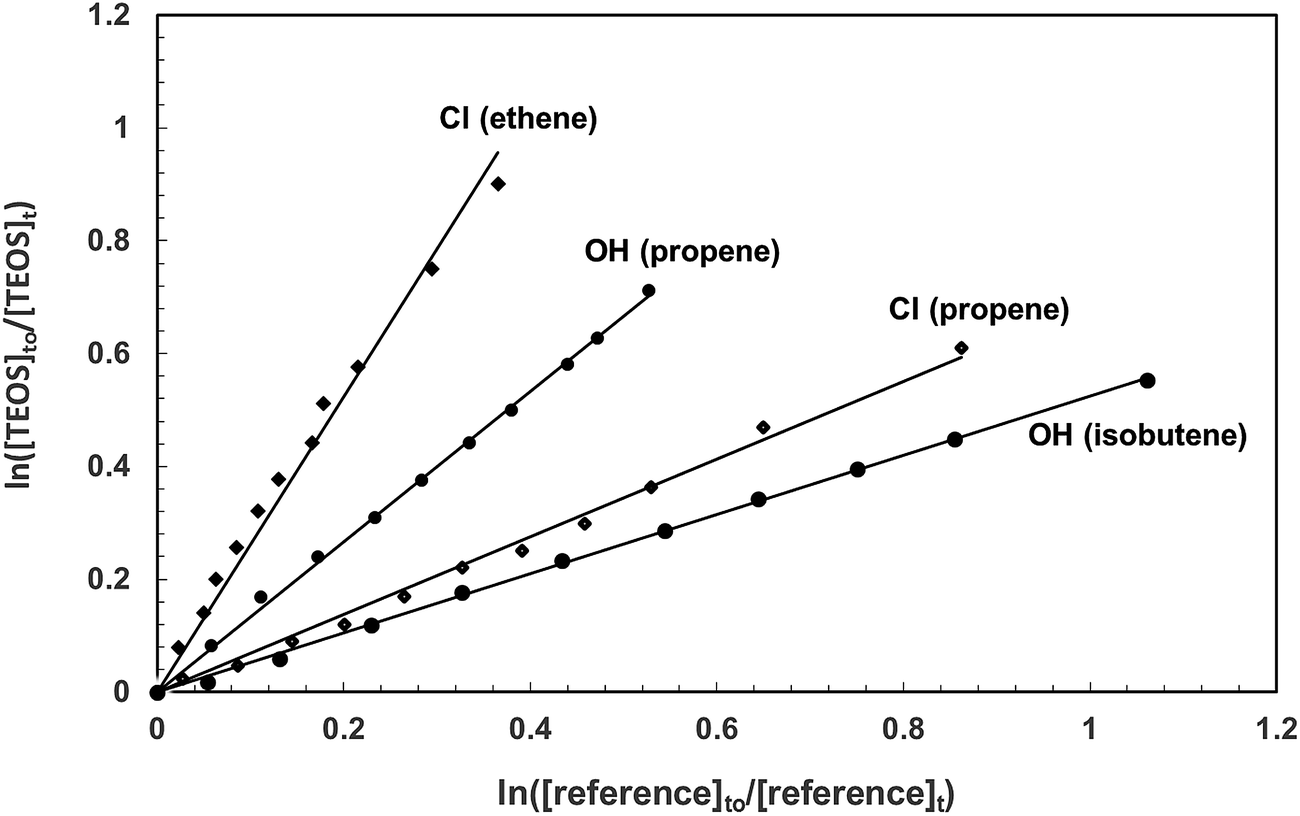 | ||
| Fig. 1 Exemplary plots of the kinetic data according to eqn (I) for the reactions of OH radicals and Cl atoms with tetraethoxysilane each measured relative to two reference compounds at 298 K in one atmosphere of synthetic air. | ||
| Reaction | Reference | k TEOS/kreference | k TEOS (cm3 per molecule per s) | SAR |
|---|---|---|---|---|
| a Calculated using the SAR of Kwok and Atkinson.7 b Estimated using the SAR of Aschmann and Atkinson36 and a substituent factor F(O) of 1.45 for the ether group Calvert et al.33 c Estimated using the SAR of Aschmann and Atkinson36 and a substituent factor F(O) of 1.18 for the ether group from Notario et al.35 | ||||
| OH + TEOS | Propene | 1.214 ± 0.073 | (3.19 ± 0.19) × 10−11 | 3.33 × 10−11a |
| 1.155 ± 0.058 | (3.04 ± 0.15) × 10−11 | |||
| 1.192 ± 0.075 | (3.14 ± 0.20) × 10−11 | |||
| 1.067 ± 0.062 | (2.81 ± 0.16) × 10−11 | |||
| Average | (3.05 ± 0.36) × 10−11 | |||
| 2-Methylpropene | 0.537 ± 0.025 | (2.91 ± 0.14) × 10−11 | ||
| 0.523 ± 0.021 | (2.83 ± 0.11) × 10−11 | |||
| 0.562 ± 0.034 | (3.04 ± 0.19) × 10−11 | |||
| Average | (2.93 ± 0.26) × 10 −11 | |||
| Cl + TEOS | Ethene | 2.217 ± 0.133 | (2.44 ± 0.15) × 10−10 | 4.87 × 10−10b |
| 2.461 ± 0.125 | (2.74 ± 0.14) × 10−10 | 4.16 × 10−10c | ||
| 2.235 ± 0.112 | (2.46 ± 0.12) × 10−10 | |||
| Average | (2.60 ± 0.30) × 10 −10 | |||
| Propene | 0.879 ± 0.053 | (2.37 ± 0.14) × 10−10 | ||
| 0.795 ± 0.040 | (2.15 ± 0.11) × 10−10 | |||
| 0.809 ± 0.063 | (2.18 ± 0.18) × 10−10 | |||
| Average | (2.22 ± 0.26) × 10 −10 | |||
The rate coefficients for the reactions of OH and Cl with TEOS listed in Table 1 were put on an absolute basis using the following rate coefficient values (in cm3 per molecule per s units) for the reference reactions: 2.63 × 10−11 and 5.41 × 10−11 for the reaction of OH with propene and 2-methylpropene;19 1.1 × 10−10 and 2.7 × 10−10 for the reactions of Cl ethene and propene.20 The reference compounds have typically an associated error of Δlog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = ± 0.07.20
k = ± 0.07.20
Since the rate coefficients for the reactions of OH and Cl obtained using the two reference compounds are in good agreement we prefer to quote final rate coefficients for the reactions which are averages of the all the determinations using the two reference compounds:
| k(OH+TEOS) = (2.99 ± 0.37) × 10−11 cm3 per molecule per s |
| k(Cl+TEOS) = (2.41 ± 0.47) × 10−10 cm3 per molecule per s |
Here the quoted errors for the coefficient values encompass the error ranges of the individual values obtained with the two reference compounds and do not include a contribution to take into account uncertainties associated with the reference compounds.
There are no previous determinations for the reaction of OH with TEOS with which the value of (2.99 ± 0.37) × 10−11 cm3 per molecule per s obtained in this work can be compared. Indeed, we could not find any studies at all in the scientific literature on the reactions of OH radicals with alkoxy substituted silanes. Gas-phase kinetic studies on the reactions of organo-silicon compounds appear to have been restricted to date to studies of the reactions of OH and NO3 radicals and O3 with mainly methyl-substituted silanes.21–26
Using the structure activity relationship (SAR) estimation method for OH rate coefficients with organics of Kwok and Atkinson7 gives a rate coefficient of 3.33 × 10−11 cm3 per molecule per s for the reaction of OH with TEOS, which is in excellent agreement with the value determined here experimentally. In the SAR method of Kwok and Atkinson7 the ethoxy group in TEOS is treated as an ether group and a value of 8.4 is used as substituent factor for the “–O–” group. It should be noted that the AOPWIN module in the EPI Suite™6 uses an older value of 6.1 as substituent factor for the “–O–“ group27 which results in a lower predicted value of 2.47 × 10−11 cm3 per molecule per s for the reaction of OH with TEOS.
Using the experimental rate coefficient for the reaction of OH with TEOS a value of 8.63 × 10−12 cm3 per molecule per s can be calculated for the reactivity of each of the four ethoxy entities (CH3CH2O–) in TEOS toward OH radicals, which can be compared with a value of 8.32 × 10−12 cm3 per molecule per s predicted by the SAR of Kwok and Atkinson.7 It is interesting to compare this reactivity with that of OH toward diethyl ether. The rate coefficient for the reaction of OH with diethyl ether is 13.1 × 10−12 cm3 per molecule per s 28,29 which on simply dividing by two would suggest an OH reactivity of 6.55 × 10−12 cm3 per molecule per s for each of the ethyl groups in diethyl ether, i.e. a similar but slightly lower reactivity than that observed for the ethyl groups in TEOS. It would appear, that within the experimental uncertainties, the –OCH2CH3 entities in TEOS are approximately equally activated as those of the –OR entities in dialkyl ethers.
As for the reaction of OH with TEOS a rate coefficient for the reaction of Cl atoms with TEOS is not available in the literature. However, the value of (2.41 ± 0.47) × 10−10 cm3 per molecule per s obtained in this work for the reaction is very similar to the value of (2.58 ± 0.44) × 10−10 cm3 per molecule per s reported in the literature for the reaction of Cl atoms with diethyl ether.30,31
SARs for the estimation of rate coefficients for the reactions of Cl atoms with alkanes have been reported by several groups and are discussed in Calvert et al.32–34 These SARs have been extended to allow the estimation of rate coefficients for VOCS containing functional groups.32–34 Notario et al.35 have reported an averaged substituent factor for the ether group of F(O) = 1.18 for a Cl SAR while Calvert et al.33 report a value of 1.45. Using these F(O) values in combination with the group rate coefficients for –CH3, –CH2– and ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CH– given in Aschmann and Atkinson36 we have estimated the rate coefficient for the reaction of Cl atoms with TEOS. The F(O) value of 1.18 from Notario et al.35 gives an estimated rate coefficient of 4.16 × 10−10 cm3 per molecule per s for the reaction while the F(O) value of Calvert et al.33 results in 4.87 × 10−10 cm3 per molecule per s (Table 1). Both values of F(O) lead to an over estimation of the rate coefficient compared to the experimental value by a factor of ∼2. A value of F(O) of around 0.4 would be required for a reasonable match between the experimental and estimated values. The F(O) value reported by Notario et al.35 was an average of the F(O) values determined for a series of ethers with the F(O) values ranging from 1.96 to 0.47. This wide variation in the substituent factor F(O) for ethers reported by Notario et al.35 would seem to indicate that either the substituent effect is not constant for different molecular constellations in the ethers or there may be difficulties with the experimental determinations of the rate coefficients used to derive the substituent factor. Based on the quality of the experimental work it seems that the former reason is probably the more plausible. Both Calvert et al.33 and Notario et al.35 have both noted that the SAR for the reaction of Cl with ethers does not work very well in many cases.
CH– given in Aschmann and Atkinson36 we have estimated the rate coefficient for the reaction of Cl atoms with TEOS. The F(O) value of 1.18 from Notario et al.35 gives an estimated rate coefficient of 4.16 × 10−10 cm3 per molecule per s for the reaction while the F(O) value of Calvert et al.33 results in 4.87 × 10−10 cm3 per molecule per s (Table 1). Both values of F(O) lead to an over estimation of the rate coefficient compared to the experimental value by a factor of ∼2. A value of F(O) of around 0.4 would be required for a reasonable match between the experimental and estimated values. The F(O) value reported by Notario et al.35 was an average of the F(O) values determined for a series of ethers with the F(O) values ranging from 1.96 to 0.47. This wide variation in the substituent factor F(O) for ethers reported by Notario et al.35 would seem to indicate that either the substituent effect is not constant for different molecular constellations in the ethers or there may be difficulties with the experimental determinations of the rate coefficients used to derive the substituent factor. Based on the quality of the experimental work it seems that the former reason is probably the more plausible. Both Calvert et al.33 and Notario et al.35 have both noted that the SAR for the reaction of Cl with ethers does not work very well in many cases.
In the case of TEOS stereo-chemical factors may also possibly be playing a role which could possibly explain why there is an apparent deactivating rather than the usually expected activating effect of the “ether group” on the rate coefficient.
Product and mechanistic study
The products formed in the reaction of OH radicals and Cl atoms with TEOS have been investigated in one atmosphere of synthetic air using the photolysis of H2O2 and Cl2 as the respective OH radical and Cl atom radical sources. Since the investigations with both OH and Cl gave identical results only the results for Cl are presented here. The spectra obtained with Cl as oxidant are H2O2 and almost water free and thus more visually informative than the spectra with OH. Fig. 2, trace A, shows the infrared spectrum of TEOS in the wavelength range 4000–700 cm−1 before irradiation of a TEOS/Cl2/air reaction mixture. Fig. 2, trace B, shows the product spectrum after irradiation and subtraction of excess TEOS. A comparison of the product spectrum with the reference spectrum of acetic acid (CH3C(O)OH) shows beyond doubt that acetic is an important reaction product. Fig. 2, trace D, is the residual product spectrum obtained on subtracting CH3C(O)OH from trace C. Plots of the yield of acetic acid versus the amount of decayed TEOS gave a molar acetic acid yield of 95 ± 4%. A typical yield plot is shown in Fig. 3. Although the plots were linear over much of the duration of the experiments some upward curative was observed toward the end of the experiments indicating that oxidation of a primary product was beginning to add to the yield of acetic acid.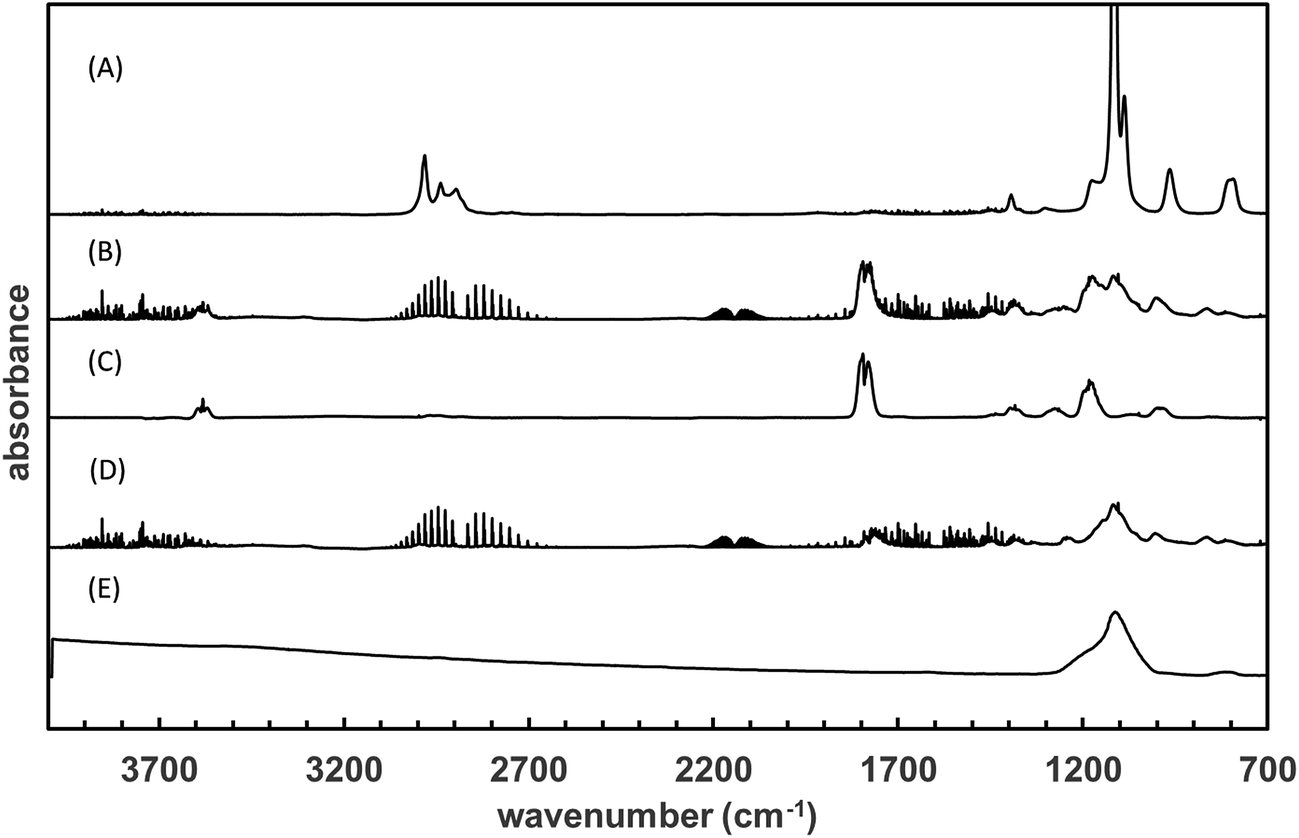 | ||
| Fig. 2 Trace (A) shows the infrared spectrum of TEOS and trace (B) shows the product spectrum obtained after irradiation of a TEOS/Cl2/air mixture. Trace (C) shows a reference spectrum of acetic acid and trace (D) shows the residual product spectrum obtained on subtraction of acetic acid from the spectrum in trace (B). Trace (E) is a reference spectrum of solid SiO2 from NIST.45 | ||
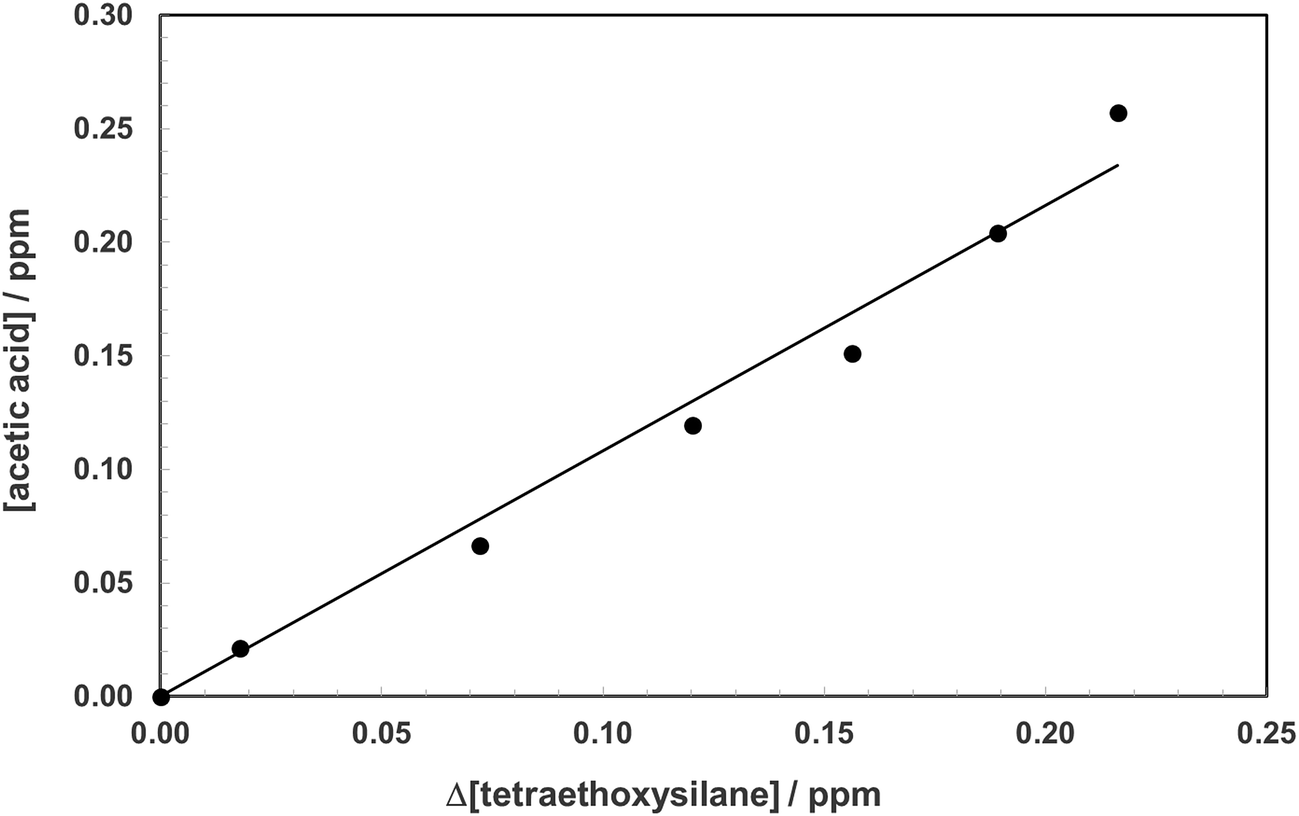 | ||
| Fig. 3 Plot of the formation of acetic acid (ppm) versus the amount of TEOS consumed in the reaction of Cl atoms with TEOS in 760 Torr of air. | ||
The molar yield of acetic acid corresponds to a carbon yield of approximately 25% for the reaction of Cl with TEOS. This result suggests that one of the ethoxy ligands is being converted quantitatively to acetic acid in the reaction. TEOS is a tetrahedral molecule:
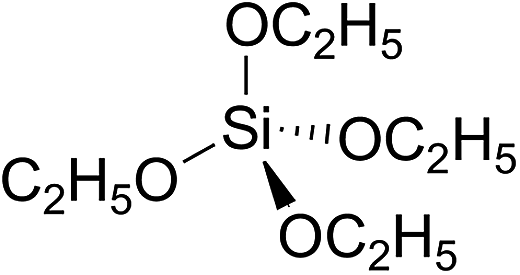
Attack of OH or Cl at one of the ethoxy groups in TEOS is expected to proceed mainly by H-atom abstraction at the –CH2– entities in the ethoxy groups:
| (C2H5O)3Si–O–CH2CH3 + OH/Cl → (C2H5O)3Si–O–C(˙)HCH3 + H2O/HCl |
The SAR of Kwok and Atkinson7 predicts only 6% H-atom abstraction at the methyl groups for the reaction of OH radicals with TEOS and we expect that for the analogue Cl reaction the contribution will not be too dissimilar. The (C2H5O)3Si–O–C(˙)HCH3 radicals will add O2 to form peroxy radicals which can undergo self-reaction and reaction with other HO2/RO2 radicals.37,38 The products of these reactions will be alkoxy radicals and carbonyl- and hydroxy-group containing products. Reaction of the peroxy radicals with HO2 could also produce a hydroperoxide.37,38
| (C2H5O)3Si–O–C(˙)HCH3 + O2 + M → (C2H5O)3Si–O–CH(OO˙)CH3 + M |
| (C2H5O)3Si–O–CH(OO˙)CH3 + RO2˙ → (C2H5O)3Si–O–CH(O˙)CH3 + RO˙ + O2 |
2(C2H5O)3Si–O–CH(OO˙)CH3 + 2RO2˙ → (C2H5O)3Si–O–CH(OH)CH3 + (C2H5O)3Si–O–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH3 + ROH + R′CHO + 2O2 O)CH3 + ROH + R′CHO + 2O2 |
| (C2H5O)3Si–O–CH(OO˙)CH3 + HO2˙ → (C2H5O)3Si–O–CH(OOH)CH3 + O2 |
Possible, reaction pathways for the (C2H5O)3Si–O–CH(O˙)CH3 alkoxy radical include reaction with O2 to form (C2H5O)3Si–O–C(O)CH3 or thermal decomposition via either a C–O bond cleavage to form (C2H5O)3Si–O˙ radicals and acetaldehyde or a C–CH3 bond cleavage to form (C2H5O)3Si–CH(O) and CH3˙ radicals.
| (C2H5O)3Si–O–CH(O˙)CH3 + O2 → (C2H5O)3Si–O–C(O)CH3 + HO2 |
| (C2H5O)3Si–O–CH(O˙)CH3 + Δ → (C2H5O)3Si–O˙ + CH3CHO |
| (C2H5O)3Si–O–CH(O˙)CH3 + Δ → (C2H5O)3Si–CH(O) + CH3˙ |
However, none of these pathways lead to the formation of acetic acid which has been established as a major product. Both of the decomposition pathways can be excluded since (i) formation of acetaldehyde was not observed and (ii) the further reactions of the CH3˙ radical would lead to the formation of HCHO and CH3OH both of which were also not observed. In fact the lack of formation of acetaldehyde and HCHO in the reaction system would support that, apart from the pathway that converts one of the ethoxy groups on TEOS to acetic acid, none of the other ethoxy groups are being detached from the Si atom in the subsequent oxidation chemistry.
We propose that the pathway leading to acetic acid formation probably involves a 1,2-hydride shift, i.e. a shift of the H-atom on the methylene carbon to the O-atom binding the (CH3CH2O)3Si˙ and ethyl entities:
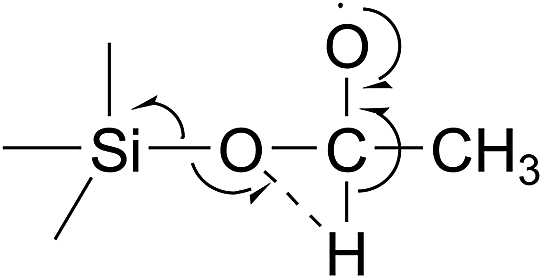
This is then followed by decomposition to form acetic acid and triethoxysilyl (C2H5O)3Si˙ radicals:
| (C2H5O)3Si–O–CH(O˙)CH3 + Δ → (C2H5O)3Si˙ + CH3C(O)OH |
After subtraction of absorptions due to acetic acid, the residual product spectrum (Fig. 2, trace D) is dominated by a large board absorption in the fingerprint region with a maximum at ∼1117 cm−1 and a few much weaker absorptions at 998, 860 and 808 cm−1. The absorption at ∼1117 cm−1 is most probably due to vibrations from a compound(s) containing a Si–O entity. A comparison is made with a reference spectrum of solid silicon dioxide (SiO2) in Fig. 2, trace E, in which a high degree of similarity is obvious.
There are very weak absorptions in the 3000 to 2800 cm−1 and 1800–1720 cm−1 regions indicating the possible presence of a product or products containing C–H and carbonyl groups. There is also a very broad weak absorption stretching from 3500 to 3200 cm−1. Such very broad bands in this region are often indicative of condensed phased H-bonded OH or aerosol formation.39
Conventionally one would assume that the fate of any triethoxysilyl radicals formed in the reaction system will be overwhelmingly addition of O2 to form the corresponding peroxy radicals.
| (C2H5O)3Si˙ + O2 + M → (C2H5O)3Si–O2˙ + M |
However, in a study on the reaction of H3Si˙ radicals with O2 evidence has been presented for the direct formation of H3SiO˙ radicals40 and a similar pathway may be possible for the reaction of O2 with (C2H5O)3Si˙ radicals.
| (C2H5O)3Si˙ + O2 + M → (C2H5O)3Si–O˙ + O˙ + M |
The near 100% molar yield of acetic acid suggests that the levels of HO2 and other RO2 peroxy radicals formed in the reaction system will be quite low and that the main fate of the (C2H5O)3Si–O2˙ radicals co-formed with acetic acid will be mainly self-reaction to form triethoxysilyloxy radicals or a molecular product:
| 2(C2H5O)3Si–O2˙ + M → (C2H5O)3Si–O˙ + RO˙ + O2 |
| 2(C2H5O)3Si–O2˙ + M → (C2H5O)3Si–O–Si(OC2H5)3 + O2 + M |
The lack of any strong C–H stretching bands in the 3000–2800 cm−1 region in the product spectrum, which would be expected for a compound such as (C2H5O)3Si–O–Si(OC2H5)3, suggests either that the compound is not formed or does not remain in the gas phase. The triethoxysilyloxy radical is a tertiary substituted radical with no α-hydrogens. It is known for analogous carbon centred radicals that the only viable atmospheric process is thermal decomposition since reaction with O2 is not possible.32 For the (C2H5O)3Si–O˙ radical this would involve ejection of an ethoxy group and formation of diethoxysilanone which is known to be very reactive.
| (C2H5O)3Si–O˙ + Δ → (C2H5O)2SiO + C2H5O˙ |
However, the further reactions of the ethoxy radicals with O2 will form acetaldehyde.
| C2H5O˙ + O2 → CH3CHO + HO2 |
Since formation of acetaldehyde was not observed in the reaction system, the contribution of the above decomposition pathway can be confidently concluded to be negligible.
Formation of acetic acid has been observed in atomic oxygen-induced Chemical Vapor Decomposition (CVD) chamber studies on TEOS/O3 using FTIR spectroscopy.41,42 Other such CVD chamber studies have used GC/MS to examine the products.43,44 These types of study are performed at elevated temperatures and the pathway generally attributed to the formation of acetic acid involves decomposition of (C2H5O)3Si–O–CH(O˙)CH3 alkoxy radicals to form triethoxysilyloxy radicals and acetaldehyde which were assumed to further oxidise in the reaction system to acetic acid.
| (C2H5O)3Si–O–CH(O˙)CH3 + Δ → (C2H5O)3Si–O˙ + M + CH3CHO |
In our study of the OH-radical initiated oxidation of TEOS we can rule out the occurrence of this pathway for the formation of acetic acid at room temperature. We suggest that the direct pathway leading to acetic formation proposed for the OH reaction may also be occurring in the in atomic oxygen-induced Chemical Vapor Decomposition (CVD) chamber studies on TEOS/O3 performed at higher temperatures.
Apart from SiO2, in CVD chamber studies formation of quite a variety of low-molecular weight products and also linear and cyclic oligomers has been observed.43 We suspect that in the OH-radical mediated oxidation of TEOS presented here many of the types of products observed in the CVD chamber studies are probably also being formed. However, as mentioned above the lack of any strong absorption features in the infrared product spectrum, apart from the strong absorption centred at 1117 cm−1 which is assigned to a compound(s) containing a Si–O entity, it would appear most of the products resulting from the further reactions of the triethoxysilyl radical under the reaction conditions of the present work (i) do not remain in the gas phase and (ii) the ethoxy groups remain attached to Si atom in the ensuing gas-phase chemistry. Positive identification of the products resulting from the reactions of the triethoxysilyl radical in air will only be possible by application of either direct analysis by an atmospheric pressure ionisation mass spectroscopy method or by product trapping and subsequent analysis by gas chromatography methods.
Acknowledgements
The authors thank Hüttenes-Albertus Chemische Werke GmbH, Düsseldorf, Germany for suppling the sample of tetraethoxysilane used in the experiments.References
- L. Rösch, P. John and R. Reitmeier, Silicon Compounds, Organic, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002, DOI:10.1002/14356007.a24_021.
- D. A. P. Bulla and N. I. Morimoto, Thin Solid Films, 1998, 334, 60–64, DOI:10.1016/s0040-6090(98) 01117–1.
- R. H. Perry and D. Green, Perry's Chemical Engineer's Handbook. Physical and Chemical Data, McGraw-Hill, New York, NY, 6th edn, 1984 Search PubMed.
- T. F. Bidleman, Environ. Sci. Technol., 1988, 22, 361–367 CrossRef CAS.
- http://toxnet.nlm.nih.gov/cgi-bin/sis/search/r?dbs+hsdb:@term+@rn+@rel+78-10-4 .
- USEPA, AOPWIN Model in Estimation Programs Interface Suite™ for Microsoft® Windows, v. 4.11, United States Environmental Protection Agency, Washington, DC, USA, 2012 Search PubMed.
- E. S. C. Kwok and R. Atkinson, Atmos. Environ., 1995, 29, 1685–1695 CrossRef CAS.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastidge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353–356 CrossRef CAS.
- W. C. Keene, A. A. P. Pszenny, D. J. Jacob, R. A. Duce, J. N. Galloway, J. J. Schultz-Tokos, H. Sievering and J. F. Boatman, Global Biogeochemistry Cycles, 1990, 4, 407–430 CrossRef CAS.
- B. J. Finlayson-Pitts, Res. Chem. Intermed., 1993, 19, 235–249 CrossRef CAS.
- C. George, W. Behnke and C. Zetzsch, ChemPhysChem, 2010, 11, 3059–3062 CrossRef CAS PubMed.
- J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander and S. S. Brown, Nature, 2010, 464, 271–274 CrossRef CAS PubMed.
- L. H. Mielke, A. Fugeson and H. D. Osthoff, Environ. Sci. Technol., 2011, 45, 8889–8896 CrossRef CAS PubMed.
- H. D. Osthoff, J. M. Roberts, A. R. Ravishankara, E. J. Williams, B. M. Lerner, R. Sommariva, T. S. Bates, D. Coffman, P. K. Quinn, J. E. Dibb, H. Stark, J. B. Burkholder, R. K. Talukdar, J. Meagher, F. C. Fehsenfeld and S. S. Brown, Nat. Geosci., 2008, 1, 324–328 CrossRef CAS.
- G. J. Philips, M. J. Tang, J. Thieser, B. Brickwedde, G. Schuster, B. Bohn, J. Lelieveld and J. N. Crowley, Geophys. Res. Lett., 2012, 39, L10811, DOI:10.1029/2012gl051912.
- C. J. Young, R. A. Washenfelder, J. M. Roberts, L. H. Mielke, H. D. Osthoff, C. Tsai, O. Pikelnaya, J. Stutz, P. R. Veres, A. K. Cochran, T. C. VandenBoer, J. Flynn, N. Grossberg, C. L. Haman, B. Lefer, H. Stark, M. Graus, J. de Gouw, J. B. Gilman, W. C. Kuster and S. S. Brown, Environ. Sci. Technol., 2012, 46, 10965–10973 CrossRef CAS PubMed.
- J. Liao, L. Gregory Huey, Z. Liu, D. J. Tanner, C. A. Cantrell, J. J. Orlando, F. M. Flocke, P. B. Shepson, A. J. Weinheimer, S. R. Hall, K. Ullmann, H. J. Beine, Y. Wang, E. D. Ingall, C. R. Stephens, R. S. Hornbrook, E. C. Apel, D. Riemer, A. Fried, R. L. Mauldin III, J. N. Smith, R. M. Staebler, J. A. Neuman and J. B. Nowak, Nat. Geosci., 2014, 7, 91–94 CrossRef CAS.
- I. Barnes, K. H. Becker and N. Mihalopoulos, J. Atmos. Chem., 1994, 18, 267–289 CrossRef CAS.
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- R. R. Baldwin, C. J. Everett and R. W. Walker, Trans. Faraday Soc., 1968, 64, 2708–2722 RSC.
- R. Atkinson, Environ. Sci. Technol., 1991, 25, 863–866 CrossRef CAS.
- R. Sommerlade, H. Parlar, D. Wrobel and P. Kochs, Environ. Sci. Technol., 1993, 27, 2435–2440 CrossRef CAS.
- E. C. Tuazon, S. M. Aschmann and R. Atkinson, Environ. Sci. Technol., 2000, 34, 1970–1976 CrossRef CAS.
- A. Goumri, J. Yuan, E. L. Hommel and P. Marshall, Chem. Phys. Lett., 2003, 375, 149–156 CrossRef CAS.
- H. Zhang, G.-L. Zhang, Y. Wang, X.-Y. Yu, B. Liu, J.-Y. Liu and Z.-S. Li, Theor. Chem. Acc., 2008, 119, 319–327 CrossRef CAS.
- R. Atkinson, Int. J. Chem. Kinet., 1987, 19, 799–828 CrossRef CAS.
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- A. Mellouki, G. Le Bras and H. Sidebottom, Chem. Rev., 2003, 103, 5077–5096 CrossRef CAS PubMed.
- P. McLoughlin, R. Kane and I. Shanahan, Int. J. Chem. Kinet., 1993, 25, 137–149 CrossRef CAS.
- L. Nelson, O. Rattigan, R. Neavyn, H. Sidebottom, J. Treacy and O. J. Nielsen, Int. J. Chem. Kinet., 1990, 22, 1111–1126 CrossRef CAS.
- J. G. Calvert, R. G. Derwent, J. J. Orlando, G. S. Tyndall and T. J. Wallington, Mechanisms of Atmospheric Oxidation of the Alkanes, Oxford University Press, Oxford, 2008 Search PubMed.
- J. G. Calvert, A. Mellouki, J. J. Orlando, M. J. Pilling and T. J. Wallington, The Mechanisms of Atmospheric Oxidation of the Oxygenates, Oxford University Press, New York, 2011 Search PubMed.
- J. G. Calvert, J. J. Orlando, W. R. Stockwell and T. J. Wallington, The Mechanisms of Reactions Influencing Atmospheric Ozone, Oxford University Press, New York, 2015 Search PubMed.
- A. Notario, A. Mellouki and G. Le Bras, Int. J. Chem. Kinet., 2000, 32, 105–110 CrossRef CAS.
- S. M. Aschmann and R. Atkinson, Int. J. Chem. Kinet., 1995, 27, 613–622 CrossRef CAS.
- P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat and F. Zabel, Atmos. Environ., Part A, 1992, 26, 1805–1964 CrossRef.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, J. Phys. Chem., 1995, 89, 1752–1755 CrossRef.
- M. Koshi, A. Miyoshi and H. Matsui, J. Phys. Chem., 1991, 95, 9869–9873 CrossRef CAS; H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, J. Phys. Chem., 1985, 89, 1752–1755 CrossRef.
- T. K. Whidden, S. Y. Lee, X. Bao, M. Couturier, J. Taylor, P. Lu, S. Romet and Z. Xiaozhong, in Proceedings of the Symposium on Fundamental Gas-Phase and Surface Chemistry of Vapor-Phase Materials Synthesis, PV98-28, ed. M. D. Allendorf, M. R. Zachariah, L. Mountziaris and A. H. McDaniel, The Electrochemical Society, 1998, pp. 153–162 Search PubMed.
- T. K. Whidden and S. Y. Lee, Electrochem. Solid-State Lett., 1999, 2, 527–530 CrossRef CAS.
- A. M. Wróbel, A. Walkiewicz-Pietrzykowska, M. Stasiak and J. Kulpiński, Chem. Vap. Deposition, 1996, 2, 285–291 CrossRef.
- T. Satake, T. Sorita, H. Fujioka, H. Adachi and H. Nakajima, Jpn. J. Appl. Phys., 1994, 33, 3339–3342 CrossRef CAS.
- http://webbook.nist.gov/cgi/cbook.cgi?Formula=SiO2%26NoIon=on%26Units=SI%26cIR=on .
| This journal is © The Royal Society of Chemistry 2016 |