
Stabilizing polysulfide-shuttle in a Li–S battery using transition metal carbide nanostructures†
Hesham Al Salema,
Venkateswara Rao Chitturia,
Ganguli Babua,
Juan A. Santanab,
Deepesh Gopalakrishnana and
Leela Mohana Reddy Arava*a
aDepartment of Mechanical Engineering, Wayne State University, Detroit, MI 48202, USA. E-mail: leela.arava@wayne.edu
bDepartment of Chemistry, University of Puerto Rico, Cayey, PR 00736, USA
First published on 11th November 2016
Abstract
Stabilizing ‘polysulfide-shuttle’ is one of the key bottlenecks that hinders the practical application of high energy density Li–S batteries. Traditional strategies to physically/chemically confine polysulfides within the pores of carbonaceous materials have resulted in limited success. In search of alternative mechanisms, herein, we present an intrinsic adsorption strategy to address both cycle life and rate capability issues in Li–S batteries. Combining experimental studies and spin-polarized Density Functional Theory (DFT) calculations, we have shown that nanostructured transition metal carbides (TMC) such as TiC and WC can adsorb polysulfides thereby enhancing the polysulfides' redox process. The combination of preferential surface adsorption of intermediate polysulfides and the reversible binding properties of lower order polysulfides on sulfiphilic metal carbide surfaces has resulted in a reversible capacity of 860 mA h g−1 and stability over 100 charge/discharge cycles. Thus, nanostructured metal carbides with their exceptional adsorption capability towards dissolved polysulfides, could be an alternative electrode candidate for Li–S batteries.
1 Introduction
Electrochemical energy storage (EES) systems with high energy density and long-term cycling performance need to be developed to meet global energy demands. Rechargeable Li–S batteries are one of the most promising EES systems and have garnered unprecedented interest due to its high theoretical capacity (1675 mA h g−1), high energy density (2500 W h kg−1), low cost, safety and environmental benignity.1–4 Additionally, the theoretical gravimetric energy density of Li–S batteries is five times higher than extant rechargeable Li-ion batteries. Li–S batteries have been investigated for many years and progress made in the recent years shows the applicability for device applications, ranging from portable electronics to electrical vehicles. However, widespread commercialization of Li–S batteries is hindered due to the practical issues; specifically, poor adsorption of soluble polysulfides (Li2Sn, n = 3 to 8) on the cathode surface and a loss of electroactive mass during the discharge/charge processes.3–6Building better Li–S batteries by overcoming the aforementioned limitations relies on the strategic development of cathode materials. In recent years, different kinds of electrode materials including nanostructured carbon–porous carbon,7–10 carbon nanotubes,11,12 carbon nanofibers,13,14 hollow porous carbon spheres,15,16 graphene,17–19 functionalized carbons,20 graphene oxide21–23 and polymer-grafted porous carbon;24,25 metal oxides–SiO2,26 TiO2,27 Al2O3,28 MnO2 (ref. 29) and Ti4O7;30 metal nitride like TiN;31 metal sulfides–CoS2,32 TiS2 and VS2;33 MXene phase Ti2C;34 polymers35,36 and very recently metal carbide like TiC37 have been investigated. According to the studies, the poor adsorption properties of hydrophobic carbon materials towards polar natured polysulfides worsen the cycling performance due to the segregation of end-discharge products on the cathode surface. Surface-functionalized carbons and polymer-grafted carbons bind favorably to the polysulfides and improve the capacity, but the overall electronic conductivity and the binding nature over the course of discharge and prolonged cycling are questionable. Metal oxides, metal sulfides and MXene phase metallic carbides chemically bind to polysulfides and facilitate multi-step redox processes in Li–S battery system. However, retention in capacity after several discharge–charge cycles and rate capability need to be improved further. Although the basic principles of sulfur electrochemistry and processes occurred in Li–S battery are known, the formulation of an efficient cathode satisfying the desired characteristics still remains elusive due to the large number of possible redox steps within the ambit. The search for an efficient cathode material is still continuing.
Numerous scientific and engineering approaches have been implemented and have directed the research to develop cathodes to improve the performances. In our previous reports, we followed a novel approach and demonstrated the utilization of nanostructured Pt and Ni electrocatalytic materials to trap polysulfides for improving the Li–S battery performance.38,39 In the present work, we have investigated the feasibility of using a new class of cathodes; specifically, transition metal carbides in Li/dissolved polysulfide battery configuration. Transition metal carbides have been used as catalysts for many electrochemical reactions including hydrogen evolution, oxygen reduction, H2O2 reduction, industrially important desulfurization, isomerization, and hydrogenolysis processes due to their unique physicochemical properties and Pt-like behavior. The electrochemical activities of transition metal carbides are found to be related to the 3d electron number of transition metal atoms and strong interactions between metal and electroactive species. Though transition metal oxides (TMOs) exhibit similar physico-chemical properties, TMCs display unique surface reactivity and catalytic properties resembling those of noble metals due to the electron distributions in carbides.40–42 In contrast to metal–oxygen bonding in TMOs, the hybrid interactions between metal d-electrons and carbon sp-electrons in TMCs causes the expansion of metal–metal distance (or) contraction of the metal d-band. As a result, d-band contraction would give a greater density of states (DOS) near the Fermi level. Thus TMCs are anticipated to exhibit excellent electron transfer characteristics thereby giving better electrochemical properties. We anticipate the characteristics of transition metal carbides such as high metallic conductivity, high work function, chemisorption of sulfur species via metal–sulfur interactions, redox ability, and excellent thermal/mechanical stability will help to improve polysulfide conversion kinetics as well as battery cycling performances. Nanostructured tungsten carbide (WC) and titanium carbide (TiC) powders were synthesized and characterized. Electrochemical studies along with ex situ microscopic/spectroscopic studies were carried out to investigate discharge/charge processes and physical insights about the interactions between metal carbide and polysulfides were provided in detail. First-principle calculations were also carried out to understand the adsorption and redox behavior of metal carbides towards lithium polysulfides.
2 Experimental section
2.1. Synthesis of transition metal carbides
In a typical synthesis of titanium carbide (TiC), 1.6 g titanium dioxide, 2.43 g magnesium carbonate and 2.92 g metallic magnesium powders were mixed thoroughly and loaded into a 50 mL capacity stainless steel autoclave. All the manipulations were carried out in a dry glove box. After sealing under an inert gas atmosphere, the autoclave was heated from room temperature to 823 K at a heating rate of 10 K min−1, and maintained at 823 K for 10 h, followed by cooling gradually to room temperature in the furnace. The product obtained from the autoclave was washed several times with dilute HCl and distilled water to remove the impurities. Finally, the product was washed with absolute ethanol and vacuum-dried at 333 K for 12 h.In a typical synthesis of tungsten carbide (WC), 6.5 g ammonium paratungstate (NH4)6W7O24 was dissolved in 300 mL of de-ionized water and followed by adding 1.8 g of activated carbon (Vulcan XC-72R, Cabot Corporation). This mixture was stirred vigorously at 343 K for 2 h to complete the wetting of carbon and impregnation of tungsten ions. Then the solvent was quickly evaporated at 373 K under continuous stirring. The resulting solid was further dried at 343 K for 6 h in a vacuum oven and then calcinated at 723 K for 2 h. In succession, this solid was carbonized at 1223 K with a heating rate of 5 K min−1 under a CO stream (flow rate: 180 to 200 mL min−1) and maintained at 1223 K for 8 h. After slow cooling to room temperature, the final product was obtained.
2.2. Preparation of lithium polysulfides (catholyte), electrodes and cell assembly
The 600 mM of electro-active species containing catholyte solution has been used to evaluate the electrochemical properties of as synthesized electrode materials. Such solution was prepared using calculated amounts of Li2S and S to attain a nominal formula of long-chain LiPS (Li2S8) in tetraethylene glycol dimethyl ether (TEGDME) at 90 °C for 24 h with effective stirring. The molar concentration is calculated based on the amount of active species i.e. sulfur in LiPS solution and the 10 μL of catholyte solution used per cell.The positive electrodes were processed by thoroughly mixing metal carbides with conductive carbon (Super-P) and polyvinylidene fluoride binder in the weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 individually. The slurry was prepared using N-methyl-2-pyrrolidone (NMP) as a solvent and coated uniformly on aluminum foil about 20 μm thickness using a doctor blade. The resulted electrodes were subjected to vacuum dry at 80 °C to evaporate NMP and used as electrodes with circular discs of 12.7 mm diameter. Standard CR2032 type coin cells were used to perform the electrochemical measurements of metal carbides as working electrodes and lithium metal as counter and reference electrodes. 10 μL of 600 mM molar concentration of catholyte solution was used as the active material along with a blank electrolyte (without polysulfides) consisting of 1 M of lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and lithium nitrate (LiNO3) in TEGDME and Celgard separator.
:10:10 individually. The slurry was prepared using N-methyl-2-pyrrolidone (NMP) as a solvent and coated uniformly on aluminum foil about 20 μm thickness using a doctor blade. The resulted electrodes were subjected to vacuum dry at 80 °C to evaporate NMP and used as electrodes with circular discs of 12.7 mm diameter. Standard CR2032 type coin cells were used to perform the electrochemical measurements of metal carbides as working electrodes and lithium metal as counter and reference electrodes. 10 μL of 600 mM molar concentration of catholyte solution was used as the active material along with a blank electrolyte (without polysulfides) consisting of 1 M of lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and lithium nitrate (LiNO3) in TEGDME and Celgard separator.
2.3. DFT calculations
To explore the adsorption state of Li2S8 on metal carbides (MCs), we performed spin-polarized Density Functional Theory (DFT) calculations. We employed a W-terminated (0001) surface for WC and Ti-terminated (111) surface for TiC as initial model of the prepared hexagonal phase WC, and cubic phase TiC. The lattice constants ware kept fixed at the experimental values: a = 2.906 Å and c = 2.837 Å for WC and a = 4.33 Å for TiC. We employed (4 × 4) surface models with 4 WC (or TiC) atomic layers and over 10 Å of vacuum space. Calculations were carried out employing the Vienna Ab initio Simulation Package (VASP).43–45 The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was employed for geometrical optimizations.46 Ionic cores were represented by the Projector Augmented-Wave (PAW) method.47,48 The wave function energy cut-off was 500 eV. A (3 × 3 × 1) Monkhorst–Pack k-point mesh was employed. Atomic positions were allowed to relax until residual forces on all atoms were less than 0.02 eV Å−1. Optimized structures of the Li2S8 molecule on these surfaces are shown in Fig. 3. As a reference system, we also performed calculations for Li2S8 on graphene. The interaction energy of Li2S8 on the MC surfaces is defined as Eint = (EMC + ELi2S8) − ELi2S8@MC, where ELi2S8@MC, EMC and ELi2S8 are the total energy of the combined systems, the MC surface and the Li2S8 molecule, respectively. Positive values of Eint indicate a favorable adsorbate–surface interaction. To determine the interaction energies accurately, we evaluated the total energies adding dispersion correction.49,502.4. Characterization
A Rigaku Miniflex II X-ray diffractometer was used to record X-ray diffraction (XRD) patterns for as synthesized metal carbide nanoparticles using a CuKα source at a scan rate of 0.02° s−1. Morphology, particle size features (field emission electron microscopy images) and elemental compositions with mapping (energy dispersive X-ray spectrum, EDX) were analyzed by JEOL JSM-7500F system operated with accelerating voltage 20 kV. Bio-logic Science Instrument potentiostat (VMP3) has been used to measure cyclic voltammograms (CV) at a scan rate of 0.1 mV s−1 in the potential window of 1.5–3.0 V. Electrochemical charge–discharge studies were performed for metal carbide electrodes at a current rate of 0.2C rate in the working potential range of 1.5–3.0 V using ARBIN charge–discharge cycle life tester.3 Results and discussion
Structural features of the prepared metal carbides were analyzed using powder XRD. Diffraction patterns of TiC and WC were indexed and presented in Fig. 1a and d, respectively. Analysis of the Bragg peaks reveal hexagonal WC phase with space group, P63mc (no. 186) and cubic TiC phase with space group, Fd3c (no. 225). Broad diffraction peaks are indicative of nanocrystalline nature. No peaks corresponding to the metal oxides or amorphous carbon were noticed, indicating phase purity of the materials. Morphological features of the metal carbides were examined using FESEM and TEM. | ||
| Fig. 1 Structural and morphological properties: (a and d) XRD, (b and e) SEM, and (c and f) TEM of TiC and WC. First and second rows correspond to the TiC and WC, respectively. | ||
SEM images depicted the agglomeration of crystallites in the size range of 20–25 nm for TiC (Fig. 1b) 80–120 nm for WC (Fig. 1e). Close examination of the particles by TEM depicted a three-dimensional network of interconnected crystallites without any impurities (Fig. 1c and f). It ensures the electronic conductivity. Further, the purity of the materials was further confirmed by composition analysis using EDX spectrum and element mapping analysis indicated the presence respective elements and also the elemental distributions for all metal carbide samples which are close to their original ratios (Fig. S1 and S2†).
The adsorption capability of metal carbides (MCs) such as tungsten carbide (WC) and titanium carbide (TiC) was further evaluated and confirmed by UV/Vis spectroscopy. 10 mmol L−1 of polysulfide solution was prepared by dissolving the adsorbate, Li2S4 in 1,2-dimethoxyethane/1,3-dioxolane in a 1:1 ratio and analyzed for static adsorption studies (Fig. 2a). The large absorption band near the wavelength of ∼320 nm attribute towards the presence of Li2S4 species. For the adsorption studies, a known amount (1 mg) of MCs was added as adsorbents to the Li2S4/DME:DOI mixture (1 mL) followed by vigorous stirring for 1 hour and kept undisturbed overnight.51 The supernatant was cautiously collected for the UV/Vis absorption studies and the disappearance of the absorption peak around ∼350 nm contributes towards the significant adsorption of polysulfides onto the MCs (Fig. 2b). Further, the disappearance of the color of the dissolved Li2S4 solution indicates the strong affinity of W and Ti edge sites towards sulfur (S). This visual inspection analysis shown in the inset of Fig. 2 shows the better adsorption property of Ti towards polysulfides which supports the electrochemical performance of MCs.
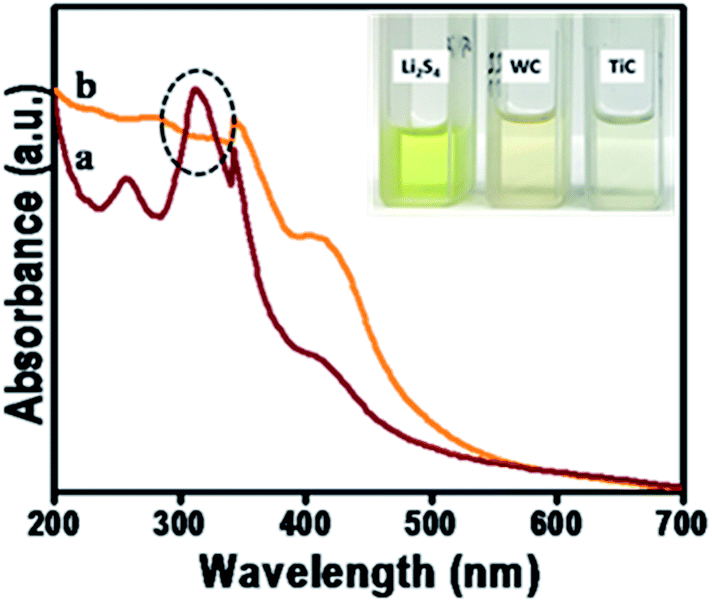 | ||
| Fig. 2 Polysulfide adsorption behavior: UV-Vis absorption spectra showing the adsorption of polysulfides onto the TiC. Inset: the visual inspection photograph of Li2S4 adsorption on WC and TiC nanostructures. | ||
Li2S8 completely dissociates upon adsorption on the MC surfaces, while it remains intact over graphene. The calculated dissociative-adsorption energy of Li2S8 on WC(0001) and TiC(111) are 3.56 eV/(S atom) and 3.68 eV/(S atom), respectively. On the other hand, the binding energy of Li2S8 on graphene is only 0.11 eV/(S atom), which agrees with previously reported52 values. WC(0001) and TC(111) showed high binding energies than graphene due to the polar metal–C bonds (Wδ+–Cδ− or Tiδ+–Cδ−) whereas graphitic carbon (C) showed low binding energy due to the non-polar C–C bonds (Fig. 3).
 | ||
| Fig. 3 DFT optimized structures of Li2S8 on TiC(111), WC(0001) and graphene. | ||
To evaluate the electrochemical performance of metal carbides such as WC and TiC, and further comparing with that of bare carbon, standard 2032 coin cells were fabricated using them as cathode vs. metallic lithium as an anode. Fig. 4 illustrates the CV of WC and TiC electrodes vs. Li/Li+ at a scan rate of 0.1 mV s−1 with 10 μL of Li2S8 in TEGDME solvent containing 1 M LiTFSI and 0.1 M LiNO3 as catholyte. Recorded CV curves for both the carbides consist of two cathodic peaks at 2.44 and 1.9 V corresponding to the transformation of long-chain lithium polysulfides to short-chain LiPS and subsequent reduction to lower lithium polysulfides respectively. Upon forward scan, anodic peak has been observed related to reversible conversion of short-chain to long-chain LiPS which results in excellent reversibility.53 Further, as a comparison between WC and TiC, the high redox peak currents for TiC indicates its stability and better activity towards polysulfide adsorption chemistry. Similarly, onset potentials for TiC have been observed are 1.91 and 2.38 V resulting in reduced polarization to that of WC and bare carbon (Fig. S3†) which is attributed to narrowly distributed TiC nanoparticles as understood from SEM images.
 | ||
| Fig. 4 Electrochemical studies: representative cyclic voltammograms of metal carbides with 600 mM of Li2S8 in TEGDME at a scan rate of 0.1 mV s−1 in the potential range of 1.5–3.0 V. | ||
To reveal the feasibility of metal carbides as electrodes towards LiPS conversion reactions, various electrochemical properties such as specific capacity, cycleability and coulombic efficiency have been studied using fabricated coin cells. The electrochemical behaviour of metal carbides was studied at a constant current rate of 0.1C (based on sulfur mass in the cell) and results have been displayed in Fig. 5. From Fig. 5, it is understood that currently synthesized nanostructured metal carbides are capable of catalyzing lithium polysulfides during charge–discharge process due to their high electronic conductivity. Both electrodes exhibit typical Li–S battery charge–discharge profiles similar to that with conventional carbon electrodes (Fig. 5a and c). However, the reduced polarization and excellent reversibility upon repeated cycling place these catalytically active electrodes a step ahead of carbon materials. Fig. 5a depicts the charge–discharge profiles of the cell containing the WC nanostructure. A stable voltage plateaus with comparable polarization between charge–discharge plateaus have been observed. The WC electrode exhibits an initial charge–discharge capacities of 720 mA h g−1 at a c-rate of 0.1C with comparable stability over 603 mA h g−1 over 100 cycles (from Fig. 5b), which reveals robustness of WC electrode even after long cycling.
 | ||
| Fig. 5 Electrochemical performance of metal carbides as electrodes towards LiPS conversion reactions vs. Li/Li+ at 0.1C rate: galvanostatic charge–discharge profiles of (a) WC and (c) TiC, cycle number vs. specific capacity and coulombic efficiency plots of (b) WC and (d) TiC. | ||
Similarly, galvanostatic measurements were conducted on TiC as cathode vs. Li/Li+ with polysulfides. TiC electrode unveiled well defined typical discharge plateaus corresponding to the formation of soluble long-chain PS and their spontaneous dissociation to short-chain PS at plateaus at 2.4 and 1.97 V and reversible conversion of short-chain to long-chain LiPS plateaus at 2.34 V during the charging process. Further, TiC exhibit a specific capacity as high as 1156 mA h g−1 with reduced polarization and excellent reversibility. The retaining nature of charge–discharge plateaus at the same potential even after number of repeated cycles infers the stability and duration of nanostructured electrode. Also, we carried out the control experiment without polysulfides to know contribution of 1 M LiNO3/LiTFSI towards the capacity. The TiC electrodes showed very negligible capacity of 180 mA h g−1 for the initial discharge and then huge capacity fade was observed (Fig. S4†). Fig. 5d shows that type of electrode material plays a crucial role in favourable polysulfide conversion process with stable specific capacity (860 mA h g−1) and coulombic efficiency (99.2%), thus related electrochemical properties upon electrochemical cycling. The impedance spectra of both the TiC and WC electrodes before and after cycling are shown in the Fig. S5† confirming the less charge transfer resistance of TiC (when compared with WC) towards the better electrochemical performance.
4 Conclusions
Owing to a high electrical conductivity, polar nature attributed PS absorptivity and high work function, metal carbides are promising as efficient electrodes for advanced Li–S batteries. Herein, metal carbides such as WC and TiC were synthesized successfully via carbothermal reduction process with desired particle size of about 100 nm. DFT calculations revealed the strong interactions between metal carbides and Li2S8 as well as electron transfer between the species. Electrochemical properties of these MCs towards LiPS conversion reactions are examined by conducting galvanostatic charge–discharge studies. Among studied MCs, TiC exhibited a stable specific capacity of 860 mA h g−1 for 100 charge–discharge cycles due to their reduced crystallite size (∼25 nm) and large number of exposed active sites.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
L. M. R. A. acknowledges the support from Wayne State University faculty startup funds.Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- M. Barghamadi, A. Kapoor and C. Wen, J. Electrochem. Soc., 2013, 160, A1256–A1263 CrossRef CAS.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- D.-W. Wang, Q. Zeng, G. Zhou, L. Yin, F. Li, H.-M. Cheng, I. R. Gentle and G. Q. M. Lu, J. Mater. Chem. A, 2013, 1, 9382–9394 CAS.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., 2011, 123, 6026–6030 CrossRef.
- M. Rao, X. Song and E. J. Cairns, J. Power Sources, 2012, 205, 474–478 CrossRef CAS.
- M. Rao, W. Li and E. J. Cairns, Electrochem. Commun., 2012, 17, 1–5 CrossRef CAS.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed.
- W. Wei, J. Wang, L. Zhou, J. Yang, B. Schumann and Y. NuLi, Electrochem. Commun., 2011, 13, 399–402 CrossRef CAS.
- X. Geng, M. Rao, X. Li and W. Li, J. Solid State Electrochem., 2013, 17, 987–992 CrossRef CAS.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- L. Ji, M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. Zhang, Energy Environ. Sci., 2011, 4, 5053–5059 CAS.
- Z. Wang, S. Zhang, L. Zhang, R. Lin, X. Wu, H. Fang and Y. Ren, J. Power Sources, 2014, 248, 337–342 CrossRef CAS.
- N. Ding, Y. Lum, S. Chen, S. W. Chien, T. S. A. Hor, Z. Liu and Y. Zong, J. Mater. Chem. A, 2015, 3, 1853–1857 CAS.
- S. Evers and L. F. Nazar, Chem. Commun., 2012, 48, 1233–1235 RSC.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS PubMed.
- G. Zhou, S. Pei, L. Li, D. W. Wang, S. Wang, K. Huang, L. C. Yin, F. Li and H. M. Cheng, Adv. Mater., 2014, 26, 625–631 CrossRef CAS PubMed.
- J. Yang, J. Xie, X. Zhou, Y. Zou, J. Tang, S. Wang, F. Chen and L. Wang, J. Phys. Chem. C, 2014, 118, 1800–1807 CAS.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- W. Zhou, H. Chen, Y. Yu, D. Wang, Z. Cui, F. J. DiSalvo and H. D. Abruña, ACS Nano, 2013, 7, 8801–8808 CrossRef CAS PubMed.
- Y. Qiu, W. Li, G. Li, Y. Hou, L. Zhou, H. Li, M. Liu, F. Ye, X. Yang and Y. Zhang, Nano Res., 2014, 7, 1355–1363 CrossRef CAS.
- G. C. Li, G. R. Li, S. H. Ye and X. P. Gao, Adv. Energy Mater., 2012, 2, 1238–1245 CrossRef CAS.
- J. Guo, Z. Yang, Y. Yu, H. c. D. Abruña and L. A. Archer, J. Am. Chem. Soc., 2012, 135, 763–767 CrossRef PubMed.
- X. Ji, S. Evers, R. Black and L. F. Nazar, Nat. Commun., 2011, 2, 325 CrossRef PubMed.
- Z. W. Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- X. Han, Y. Xu, X. Chen, Y.-C. Chen, N. Weadock, J. Wan, H. Zhu, Y. Liu, H. Li and G. Rubloff, Nano Energy, 2013, 2, 1197–1206 CrossRef CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- Z. Cui, C. Zu, W. Zhou, A. Manthiram and J. B. Goodenough, Adv. Mater., 2016, 28, 6926–6931 CrossRef CAS PubMed.
- Z. Yuan, H.-J. Peng, T.-Z. Hou, J.-Q. Huang, C.-M. Chen, D.-W. Wang, X.-B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- Z. W. Seh, J. H. Yu, W. Li, P.-C. Hsu, H. Wang, Y. Sun, H. Yao, Q. Zhang and Y. Cui, Nat. Commun., 2014, 5, 5017 CrossRef CAS PubMed.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- F. Wu, J. Chen, R. Chen, S. Wu, L. Li, S. Chen and T. Zhao, J. Phys. Chem. C, 2011, 115, 6057–6063 CAS.
- L.-X. Miao, W.-K. Wang, A.-B. Wang, K.-G. Yuan and Y.-S. Yang, J. Mater. Chem. A, 2013, 1, 11659–11664 CAS.
- H.-J. Peng, G. Zhang, X. Chen, Z.-W. Zhang, W.-T. Xu, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12990–12995 CrossRef CAS PubMed.
- H. Al Salem, G. Babu, C. V. Rao and L. M. R. Arava, J. Am. Chem. Soc., 2015, 137, 11542–11545 CrossRef CAS PubMed.
- G. Babu, K. Ababtain, K. Y. S. Ng and L. M. R. Arava, Sci. Rep., 2015, 5, 8763 CrossRef CAS PubMed.
- Y. Zhong, X. Xia, F. Shi, J. Zhan, J. Tu and H. J. Fan, Adv. Sci., 2016, 3, 1500286 CrossRef PubMed.
- A. L. Ivanovskii, D. L. Novikov and V. A. Gubanov, Phys. Status Solidi B, 1987, 141, 9–33 CrossRef CAS.
- D. Ham and J. Lee, Energies, 2009, 2, 873 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- Q. Zhang, Y. Wang, Z. W. Seh, Z. Fu, R. Zhang and Y. Cui, Nano Lett., 2015, 15, 3780–3786 CrossRef CAS PubMed.
- F. Y. Fan, W. H. Woodford, Z. Li, N. Baram, K. C. Smith, A. Helal, G. H. McKinley, W. C. Carter and Y. M. Chiang, Nano Lett., 2014, 14, 2210–2218 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22434b |
| This journal is © The Royal Society of Chemistry 2016 |