
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzoxazines with enhanced thermal stability from phenolated organosolv lignin
Ghizelle Jane
Abarro†
ab,
Jacob
Podschun†
c,
Leslie Joy
Diaz
b,
Seishi
Ohashi
a,
Bodo
Saake
c,
Ralph
Lehnen
d and
Hatsuo
Ishida
 *a
*a
aDepartment of Macromolecular Science and Engineering, Case Western Reserve University, Cleveland, Ohio 44106-7202, USA. E-mail: hxi3@case.edu; Tel: +1 216 368 4285
bDepartment of Mining, Metallurgical and Materials Engineering, University of the Philippines, Diliman, Quezon City, Philippines
cDepartment of Wood Science and Chemical Wood Technology, University of Hamburg, Leuschnerstraße 91b, 21031 Hamburg, Germany
dThünen Institute of Wood Research, Leuschnerstraße 91b, 21031 Hamburg, Germany
First published on 28th October 2016
Abstract
Lignin-based benzoxazines are synthesized for the first time using organosolv lignin as the phenolic component and aniline or propargyl amine as the amine component through the Mannich condensation reaction. Acid-catalyzed phenolation of organosolv lignin is performed to increase the phenolic structure with the open ortho-position, which is a requirement for an oxazine ring formation. Two model compounds using o-cresol and p-cresol as the phenolic component and propargylamine as the amine component are also synthesized for comparison. The successful syntheses are verified by Fourier transform infrared spectroscopy (FT-IR); proton, carbon and phosphorus nuclear magnetic resonance spectroscopy (1H, 13C and 31P NMR); and elemental analysis. Further structural characterization of the precursor resins is performed using heteronuclear single quantum coherence (HSQC) NMR technique. The polymerization process is followed by both differential scanning calorimetry (DSC) and in situ isothermal FT-IR technique. The polymerization of the lignin-based benzoxazines proceeds faster than ordinary benzoxazine monomers due to the catalytic effect of the residual phenolic moieties in the lignin units. The majority of polymerization process takes place in less than 15 min at 180 °C for both lignin-based benzoxazines studied. The thermal stability of the polymers under study is evaluated by thermogravimetric analysis (TGA). The char yields of the polybenzoxazines derived from the lignin-based benzoxazines are close to 50%, which lead to LOI values considered self-extinguishing.
1 Introduction
Efforts to shift dependence from petroleum-based feedstock target the use of biofuels (bioethanol and biodiesel) as possible alternatives amongst others; yet the high production costs have rendered it underutilized. To address the lack of economical competitiveness, great focus is given to possible value-added applications. The wealth of reactive moieties in lignin's structure makes it a good candidate in the search for renewable feedstocks for polymer production.1 However, the utility of lignin remains limited owing to the variability of its molecular structure, which depends on geographic origin,2,3 phytogenetics,4 plant morphology5,6 and isolation method.7Polybenzoxazine is a recently commercialized polymer with wide applicability and favorable properties.8 When utilizing lignin for benzoxazine synthesis, a proposed method is to break down lignin into benzenes or phenols for polymer synthesis.9 One problem however, is that the products obtained were mostly mixtures of isomers and complete conversion into pure compounds is still a curiosity.10 An alternative approach is the direct conversion of the aromatic hydroxyl groups in the lignin molecule to benzoxazines. However, high contents of syringyl units and low concentrations of p-hydroxyphenyl units7 hinder conversion into high density of benzoxazines and thus prohibit an extended polybenzoxazine formation. Benzoxazine can flexibly be synthesized from almost any kind of phenolic derivatives and primary amine as long as the phenolic structure possesses an unsubstituted ortho position. However, due to this lack of phenolic structure with open ortho position, lignin was rarely considered as direct building block.11 This flexibility however appealed to the use of other renewable materials. Benzoxazine monomers have been derived from bio-based phenolic substances such as vanillin from the vanilla orchid,12 cardanol from cashew nut shell,13 terpene from orange rind,14 and urushiol from poison ivy.15 Recently, Podschun et al.16 presented the acid-catalyzed phenolation of beech organosolv lignin to yield an activated lignin with a multitude of phenolic groups having two free ortho sites (Fig. 1, step 1). By this means, lignin could function as the backbone of the benzoxazine.
 | ||
| Fig. 1 Phenolation, benzoxazine synthesis and subsequent polymerization to polybenzoxazine with intramolecular six-membered ring of hydrogen bonds. | ||
Recently, the overall compatibility of lignin with polybenzoxazines has been studied. In 2012, blends of lignin with benzoxazine resin were studied by El Mansouri et al. and Haque et al.17,18 Both groups observed that higher amounts of lignin led to lower polymerization temperatures (Tp) and higher glass-transition temperatures (Tg) but at the expense of phase homogeneity.18 Lignin also improved the char yield, an indicative parameter of flame retardance. Upon calculation of the limiting oxygen index, the blend material was classified as self-extinguishing.17 Ougi et al. filed a patent on a lignin compound, in which the phenolic structures were converted into benzoxazine moieties. Sufficient free reactive aromatic ortho positions were achieved by mixing the new compound with a phenolic resin in a blend.19 In 2013, Chiou and Ishida suggested that such method resulting in coexistence of lignin and benzoxazine on the same molecule might amplify the benefits reported by El Mansouri et al. and Haque et al.11 Later on that year, Comí et al. reported synthesis of benzoxazines from phenolic compounds that resemble the lower molecular weight substructures in lignin.20 The lack of phenolic structures with unsubstituted ortho position in naturally derived lignin decomposition products rendered this approach limited success since free ortho positions only constitute a small fraction of total phenolic structures.
In order to overcome this difficulty, this study explores the effects of forming more benzoxazine moieties in the lignin structure by using phenolated beech organosolv lignin (PL), which contained the highest values of reactive functionalities among lignins studied.21 Aniline (abbr. a, Fig. 1, step 2) was chosen as it is a commonly used amine component for most basic benzoxazines. Additionally, the more reactive propargylamine (abbr. pgl, Fig. 1, step 2), which forms a stable aromatic structure upon crosslinking, was investigated to study potential synergistic effects that may result with lignin. The thermal behavior of both lignin-based benzoxazines (aniline and propargyl-based benzoxazines are hereinafter abbreviated as PL-a and PL-pgl, respectively) was compared to two model compounds based on p- and o-cresols to resemble p- and o-monosubstituted phenol attached to lignin.
Thus the biopolymer lignin could potentially be equipped with the interesting properties of benzoxazines, namely, that they polymerize without the aid of harsh catalysts and do not produce volatile organic compounds. Furthermore, benzoxazine resins usually show near-zero volume shrinkage during polymerization, low water absorption, glass transition temperature much higher than polymerization temperature, excellent heat resistance and flame retardance, fast mechanical property development at low crosslinking levels, and very low surface energy.22–25 These advantageous properties result in various high-performance applications in fields such as aerospace, electronics, and composites.8
The abovementioned characteristics are common to all poly-benzoxazines because of the benzoxazine unit, which upon polymerization forms a six-membered ring due to intramolecular hydrogen bonds between the phenolic OH group and the nitrogen atom of the tertiary amine in polybenzoxazine (Fig. 1, step 3).
2 Experimental
2.1 Extraction and phenolation of lignin
Beech organosolv lignin was produced and phenolated according to our previous publication.16 Phenolation conditions applied included: phenol (20 g, 0.21 mol), lignin (10 g, ∼0.05 mol C9 units), H2SO4 (1.1 mL, 0.02 mol), T = 110 °C, t = 20 min. The phenolated lignin (PL) was determined to contain 4.3 mmol g−1 phenol attached to lignin.2.2 Synthesis of benzoxazines based on phenolated lignin and aniline (PL-a) or propargylamine (PL-pgl)
In a 25 mL round bottom flask, the amine (a: 786 μL, pgl: 550 μL, 8.6 mmol) was reacted with paraformaldehyde (516 mg, 17.2 mmol) in 1 mL toluene–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, by vol) for 15 min under nitrogen atmosphere at 95 °C to form a clear solution. 1 g phenolated lignin (PL) completely dissolved in 4 mL toluene–ethanol (1:1, by vol) was added. The reaction was proceeded at 95 °C under reflux and nitrogen atmosphere for 20 h. After cooling to r.t., the lignin–benzoxazines were precipitated by adding 50 mL diethyl ether. The solids were collected on cellulose acetate membrane, washed with 200 mL diethyl ether, and dried under vacuum to obtain light-brown powders in yields of 1.24 g (82%) for PL-a, and 1.00 g (78%) for PL-pgl.
:1, by vol) for 15 min under nitrogen atmosphere at 95 °C to form a clear solution. 1 g phenolated lignin (PL) completely dissolved in 4 mL toluene–ethanol (1:1, by vol) was added. The reaction was proceeded at 95 °C under reflux and nitrogen atmosphere for 20 h. After cooling to r.t., the lignin–benzoxazines were precipitated by adding 50 mL diethyl ether. The solids were collected on cellulose acetate membrane, washed with 200 mL diethyl ether, and dried under vacuum to obtain light-brown powders in yields of 1.24 g (82%) for PL-a, and 1.00 g (78%) for PL-pgl.
2.3 Synthesis of the propargylamine-based model compounds (oC-pgl and pC-pgl)
The two model compounds used in this study were 8-methyl-3-(prop-2-yn-1-yl)-3,4-dihydro-2H-benzo[e][1,3]oxazine and 6-methyl-3-(prop-2-yn-1-yl)-3,4-dihydro-2H-benzo[e][1,3]oxazine, which were derived from the reaction of propargylamine with o-cresol and p-cresol (oC-pgl and pC-pgl, respectively). The following were added in a 25 mL round bottom flask: p-cresol/o-cresol (2.42 g, 22 mmol), propargylamine (1.43 mL, 22 mmol), paraformaldehyde (1.35 g 45 mmol), and toluene (5.19 mL, 49 mmol) as solvent. The mixture was heated under reflux at 95 °C for 3 h. Both products were washed three times with 3 N NaOH to remove unreacted phenols; and three times with distilled water to remove residual NaOH in the solution. The product was dried with anhydrous sodium sulfate and filtered. Toluene was removed by air blowing for 24 h followed by drying at 50 °C in a vacuum oven for 24 h. Both were obtained as clear fluids with oC-pgl being colorless and pC-pgl having a yellowish color. The yields were 88% and 95%, respectively.
1H NMR δ (300 MHz, ppm, CDCl3): oC-pgl: 2.24 (Ar–CH3), 2.40 (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 3.62 (–CH2–), 4.14 (Ar–CH2–N), 4.98 (O–CH2–N) and 6.86–7.04 (Ar–H). pC-pgl: 2.27 (Ar–CH3), 2.33 (CH), 3.60 (–CH2–), 4.09 (Ar–CH2–N), 4.91 (O–CH2–N), and 6.72–6.97 (Ar–H).
CH), 3.62 (–CH2–), 4.14 (Ar–CH2–N), 4.98 (O–CH2–N) and 6.86–7.04 (Ar–H). pC-pgl: 2.27 (Ar–CH3), 2.33 (CH), 3.60 (–CH2–), 4.09 (Ar–CH2–N), 4.91 (O–CH2–N), and 6.72–6.97 (Ar–H).
2.4 Equipments and characterization
990 Hz spectral window, 256 scans, acquisition time 1.0 s and a 20 s delay between pulses.
000 Hz spectral window, 20000 scans, 1.4 s acquisition time and 2.0 s delay between pulses.
Signal assignments in NMR spectroscopy referred to the literature.16,30
250 g mol−1, Agilent) were applied for calibration of the RI detector (RI 71, Shodex). Sample detection was performed using an UV detector (UV-2077, Jasco) at 280 nm, and phenol red was used to match detectors. The data was recorded and evaluated using WinGPC Unichrom V8.10 software from polymer standards service.
For in situ FT-IR, the samples were heated to 180 °C by purging with dry air. Starting from room temperature, the target temperature was reached within 10 min. Spectra were recorded at 0, 5, 10, 15, 30, 60, 180 min isothermally.
3 Results and discussion
3.1 Thermal properties of phenolated organosolv lignin (PL)
The thermal properties of the phenolated lignin evaluated by DSC (Fig. 2) showed a transition at 107 °C, which was hypothesized to be a glass transition. The second transition event was observed at 251 °C. Previous studies that reported the same observation linked such events with the occurrence of softening in amorphous and semi-crystalline polymers.31 Both, glass transition and softening, were reversible phenomena and were retained in the third heating run at 107 °C and 251 °C, respectively. Since the energies absorbed were very low, they were not expected to significantly affect thermoset polymerization. | ||
| Fig. 2 DSC thermogram of phenolated lignin PL including 1st derivative of heat flow (gray). | ||
The thermogravimetric analysis in Fig. 3 shows a wide temperature range, at which PL decomposes. This is usually related to the presence of various oxygen-containing functional groups decomposing at different temperatures. Decomposition began at 297 °C, which is associated with the degradation of the propanoid portion to form methyl-, ethyl-, and vinyl-compounds.32 It continued towards a maximum decomposition temperature of 358 °C as the lignin structure was progressively broken down and weakly bonded groups volatilized.33–35 However, these degradation products were partially free radicals that either react with each other or with non-degraded lignin via radical–radical interactions or electron transfer.36,37 Such reactions result in heavier intermediate compounds and even rearrangement of the backbone to form char.38 The char yield of PL at 800 °C was 17.3%, which was slightly higher than that of the unmodified lignin material with a char yield of 15.8%. The increased char yield might be due to the higher content of aromatic structures in PL available for char formation.
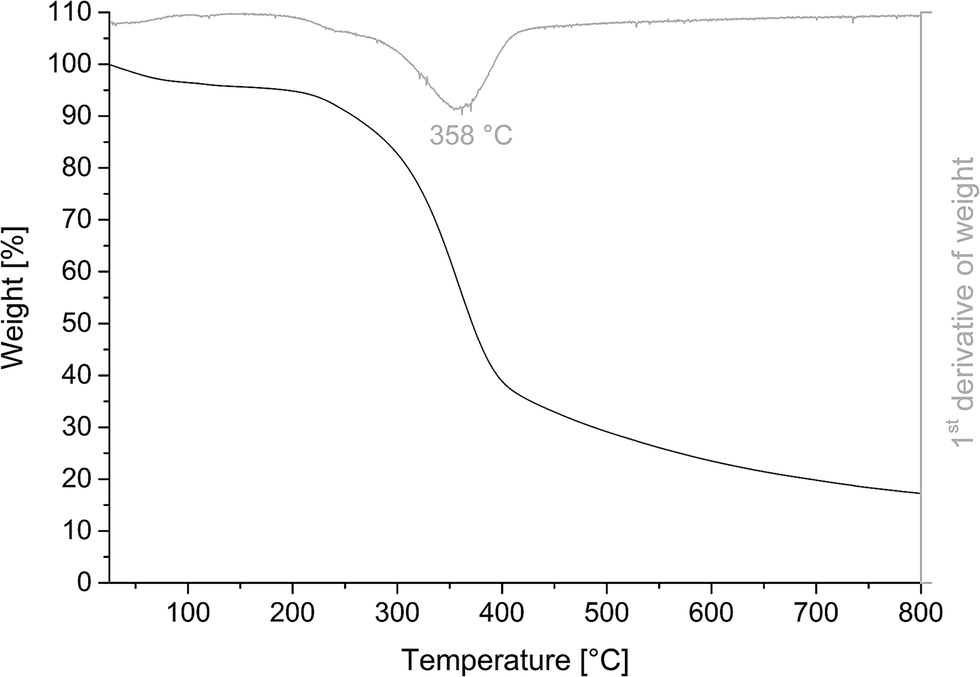 | ||
| Fig. 3 TGA thermogram of phenolated lignin PL including 1st derivative of weight (gray). | ||
3.2 Synthesis and structural characterization of lignin-based benzoxazines
The initial stage of synthesis proceeds by a low-temperature Mannich condensation of the amine component and formaldehyde. This reaction results in a hemiaminal intermediate that forms a perhydrotriazine-ring.39–41 Upon opening of the perhydrotriazine ring, it reacts with the phenols in PL to form an oxazine ring structure (Fig. 1, step 2).The hydroxyl groups of both products PL-a and PL-pgl were compared to those of PL by 31P NMR spectroscopy (Fig. 4). All signal intensities below 141.4 ppm were largely reduced. Especially, the signals at 138.0 ppm referring to para-attached phenol units in PL and at 139.6 ppm referring to guaiacyl units almost quantitatively vanished. From the original integrals of PL for ortho- and para-attached phenol and guaiacyl (4.7 mmol g−1) only 0.7 mmol g−1 for PL-a and 1.1 mmol g−1 for PL-pgl remained. It was consequently deduced that the majority of active phenolic hydroxyl groups in PL were converted to benz-oxazines. The integrals of signals for aliphatic hydroxyls, 5-substituted moieties and carboxyl groups displayed marginal reduction (PL: 0.2/1.5/0.0; PL-a: 0.1/1.2/0.0; PL-pgl: 0.2/1.3/0.0 mmol g−1 respectively), potentially related to the mass increase after benzoxazine formation. However, an additional peak at 138.8 ppm (PL-a) and 138.5 ppm (PL-pgl) was present. Being present in both products at similar chemical shifts, these peaks signified either an aromatic hydroxyl group or an amino group. However, aromatic and aliphatic amino groups (from aniline and propargylamine) show larger chemical shift separations than that observed.42 Therefore, these additional peaks were attributed to the unreacted ortho-phenols in lignin. The 31P NMR spectra indicated that in addition to the conversion of nearly all p-hydroxyphenyl-equivalent structures to benzoxazines, guaiacyl units were also converted. These results underline that benzoxazine formation proceeded via aromatic substitution to etherification/ring closure with the hydroxyl group as it appeared that only hydroxyl groups with free ortho sites reacted (not the 5-substituted units).
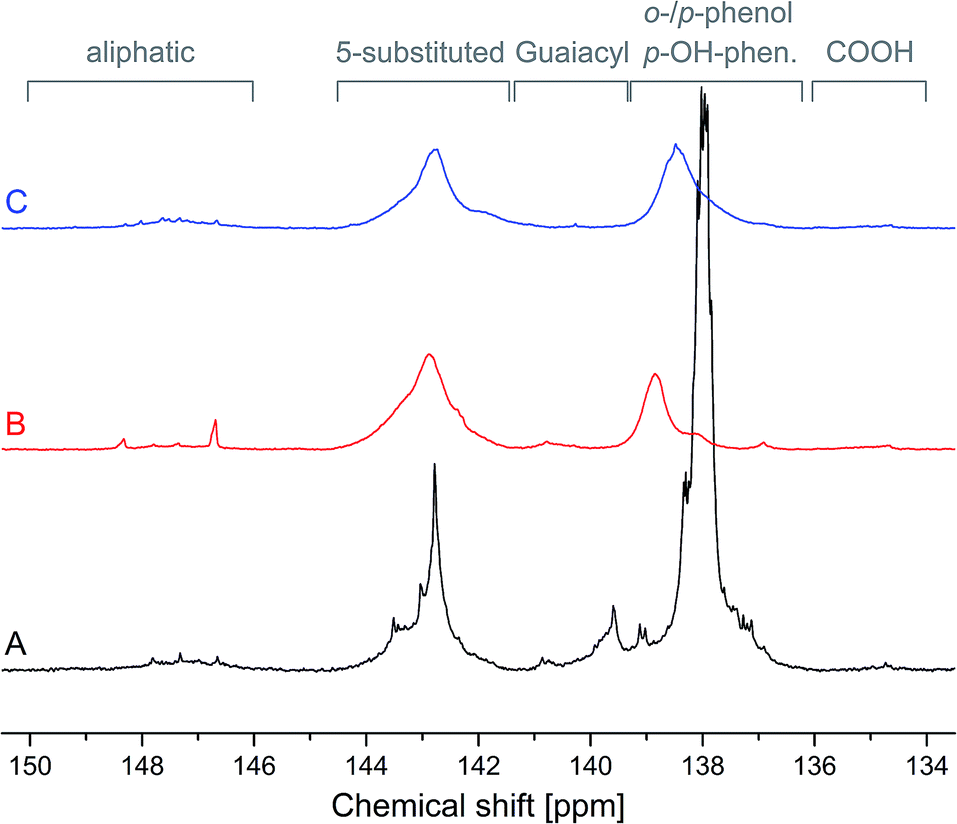 | ||
| Fig. 4 31P NMR spectra of phenolated lignin PL (A), and resulting benzoxazines PL-a (B) and PL-pgl (C) normalized on IS at 151.9 ppm. | ||
The structure of the PL-derived benzoxazines was further characterized via1H/13C HSQC NMR. The resonances for PL-pgl were found to overlap in one-dimensional NMR spectra, but could partially be resolved in HSQC spectra. In Fig. 5, the spectra for both lignin–benzoxazines were compared with the spectrum of PL. For PL-a, new signals were observed at 4.6/48.9 ppm and 5.4/78.2 ppm; and for PL-pgl at 3.9/48.6 ppm and 4.8/80.3 ppm. These peaks referred to the methylene groups in the benzoxazine ring (Ar–CH2–N and O–CH2–N, respectively).30 The separation of approximately 0.8 and 0.9 ppm between these 1H resonances was similar to many other benzoxazines in literature, where separation frequencies in the range of 0.8–0.9 ppm were reported.8 The ratio of integrated peak intensity in the spectra was 1.01 (δ4.6/48.9 ppm/δ5.4/78.2 ppm) for PL-a and 0.93 (δ3.9/48.6 ppm/δ4.8/80.5 ppm) for PL-pgl. The integrals for PL-a were verified by 13C NMR (ratio: 1.02, Fig. 6) and 1H NMR (ratio: 0.99). Integration for PL-pgl was not performed due to overlapping signals (Fig. 5 and 6). Based on these NMR analyses, the formation of the benzoxazine structures was apparent in both samples. For PL-pgl, two additional signals at 3.5/39.6 ppm and 3.2/74.9 ppm arose, representing the methylene and methylidyne group in propargylamine, respectively.30 Peaks assigned to PL side chain and methoxyl groups remained with reduced intensities.16 The cross-resonances between 6.6/106.3 ppm and 7.3/129 ppm increased for PL-a, representing aromatic C–H from aniline attached. For PL-pgl, the resonances of aromatic C–H, especially between 6.5 and 7.3 ppm, decreased due to aromatic substitution of free ortho positions.16
 | ||
| Fig. 5 1H/13C-HSQC spectra of phenolated lignin PL (A) and resulting benzoxazines PL-a (B) and PL-pgl (C) with 1H NMR traces. | ||
 | ||
| Fig. 6 13C NMR of phenolated lignin PL (A) and resulting benzoxazines PL-a (B) and PL-pgl (C). | ||
Besides the two-dimensional cross peaks, new quaternary carbons were identified in 13C NMR (Fig. 6). These included peaks at 80.1 ppm of the quaternary C in the propargyl group, at 115.7 ppm of Cphenol–CH2–N, at 151.8 ppm of C1phenol in PL-a, at 151.4 ppm of C1phenol in PL-pgl and at 147.8 ppm of C1anilin in PL-a. The original signal at 155.1 ppm of C1phenol in PL nearly quantitatively vanished in PL-a and PL-pgl.
The two model compounds are in agreement with the analytical data described by Nagai et al.,30 whose NMR chemical shifts just slightly varied in 13C NMR due to change in solvent. The amounts of benzoxazine moieties formed were estimated by 1H NMR spectroscopy with internal standard to be 2.0 mmol g−1 for PL-a and 2.1 mmol g−1 for PL-pgl using the peak at δ = 5.4 ppm and 4.8 ppm, respectively (1H traces in Fig. 5).
The incorporation of the amine compounds was examined by elemental analysis. The nitrogen contents measured were 4.5% (3.2 mmol g−1) for PL-a, and 4.1% (2.9 mmol g−1) for PL-pgl. The theoretical nitrogen content for complete conversion could be approximated based on the benzoxazines unit molar mass (PL-a: 119.2 g mol−1, PL-pgl: 81.1 g mol−1), which adds to the phenolic hydroxyl group of PL (p-hydroxyphenyl + guaiacyl: 4.3 + 0.4 mmol g−1) to yield 3.0 mmol g−1 for PL-a and 3.4 mmol g−1 for PL-pgl. Thus, theoretical and experimental values for nitrogen contents were in close agreement. However, lower values were obtained from 1H NMR quantification, indicating that some nitrogen moieties were otherwise included into the products, besides benzoxazine structures.
In the FT-IR analysis shown in Fig. 7, the lignin–benzoxazines PL-a and PL-pgl are compared to the phenolated lignin PL and two model compounds oC-pgl and pC-pgl (benzoxazines based on ortho- and para-cresol, respectively and propargylamine). In this analysis, the obvious appearance of the band around 940 cm−1 is further evidence of the benzoxazine formation as it corresponds to the out-of-plane bending of a benzene ring with an oxazine ring attached. For propargylamine-based benzoxazines, the appearance of a sharp peak around 3283 cm−1 and 645 cm−1 confirmed the attachment of acetylene groups in the structures.43,44 Traces of paraformaldehyde would also occur near this band as well, but such possibility was discarded since additional paraformaldehyde bands (e.g. at 1093 cm−1) were absent.45 Other bands in the benzoxazine spectra can be seen at 1227 cm−1 (Ar–O–C asymmetric stretch), 1029 cm−1 (Ar–O–C symmetric stretch), 1328 cm−1 (CH2 wagging of oxazine), and 2750–3050 cm−1 (CH2 stretch of methylene).8 However, these bands also occurred in PL, though with different intensities. Nevertheless, the large broad peak corresponding to phenols in PL (3150–3600 cm−1) apparently decreased for both PL-a and PL-pgl signifying the reaction of phenols to form benzoxazine moieties.8,45
 | ||
| Fig. 7 FT-IR spectra of phenolated lignin PL, benzoxazines PL-a and PL-pgl as well as model compounds oC-pgl and pC-pgl showing peaks for benzoxazines (940 cm−1) and terminal propargyl (645 and 3283 cm−1). | ||
Beyond spectroscopic parameters, the change in molecular weight distribution was also investigated (Fig. 8). The weight average molecular weights were determined to be 2200 g mol−1 for PL, 2900 g mol−1 for PL-a, and 5300 g mol−1 for PL-pgl. The molar-mass dispersities are 2.8, 2.3 and 3.0, respectively. An increase in the molecular weights of both lignin–benzoxazines is expected due to the addition of benzoxazine units in the lignin skeleton. The theoretical values, 3600 g mol−1 for PL-a and 2900 g mol−1 for PL-pgl, are derived from the calculated C9-unit weights of 340 g mol−1 for PL, 550 g mol−1 for PL-a and 450 g mol−1 for PL-pgl (C9 of plain lignin about 200 g mol−1 and 1.8 phenols per C9). The variation in the experimental values of PL-a (i.e. lower than theoretical) and PL-pgl (i.e. higher than theoretical) might be associated with different pKa values of both amines. The stronger basicity of propargylamine might have resulted in more intense condensation reactions such as with formaldehyde, whereas weak basicity might predominantly have resulted in ether cleavage.46 Alternatively, a small fraction of the propargyl units might have polymerized already during synthesis of benzoxazines.
 | ||
| Fig. 8 Molecular weight distributions of raw (dotted gray) and phenolated lignin (dashed black) as well as benzoxazines PL-a (red) and PL-pgl (blue). | ||
Overall, it can be concluded that the product characteristics were largely attributed to the formation of benzoxazines.
3.3 Polymerization behavior of lignin-based benzoxazines
The polymerization process of PL-a and PL-pgl was investigated through the in situ FT-IR spectra shown in Fig. 9, wherein characteristic absorptions of benzoxazines (around 940 cm−1 and 1227 cm−1)8 quickly disappeared, indicating the opening of benzoxazines rings. This rather fast polymerization at 180 °C is catalyzed by the existing phenolic groups acting as benzoxazine polymerization catalyst. The presence of a small amount of phenolic groups is evident by the broad bands in the range 3300–3600 cm−1 in the FT-IR spectra of PL-a and PL-pgl. | ||
| Fig. 9 In situ FT-IR spectra of PL-a (top) and PL-pgl (bottom) during the polymerization at 180 °C. | ||
In PL-pgl, the signals referring to propargyl structures (3283 cm−1 and 645 cm−1)43,44 vanished because propargyl units have also polymerized. This thermally activated reaction is reported to result in cyclotrimerization of three ethynyl end groups to form a stable benzene ring, which might be indicated by the peak at 1657 cm−1.47,48 The cyclotrimerization can lead to crosslinked structure in addition to the polymerization due to the multiple benzoxazine groups in the lignin molecule. According to Demir et al. the cyclotrimerization usually occurs around or above 220 °C.44 However, this process is known to be responsive to a wide range of catalysts. The rich functionality of lignin possibly induced a catalytic effect causing the polymerization to occur at a lower temperature, i.e. at isothermal heating temperature of 180 °C. From the in situ FT-IR spectra in Fig. 9 it can be seen that the polymerization of PL-a continued for 30 min, and of PL-pgl for 60 min. In both samples, only minor changes occurred afterwards until 180 min.
The polymerization process of lignin–benzoxazines and model benzoxazine compounds was additionally monitored by DSC (Fig. 10), and compared to BA-a in Table 1, a basic benzoxazine structure widely regarded as the standard benzoxazine monomer, which is derived from bisphenol-A and aniline. BA-a was used previously with regard to benzoxazine–lignin blends.17 By directly using lignin to synthesize the benzoxazine PL-a, the onset and maximum polymerization temperatures were similarly low as compared to the blend of BA-a and lignin reported by El Mansouri et al.17 In the previous studies, it was recognized that acidic hydroxyl groups, which were also present in PL, catalyzed the polymerization processes.49 However, the catalytic effect was more prominent for blends than lignin-based benzoxazines. This observation could be attributed to the reduction of hydroxyl groups in the latter during formation of oxazine rings (except 5-substituted units).
 | ||
| Fig. 10 DSC thermograms of lignin–benzoxazines and model compounds (y-axes for the two sets were scaled differently, see Table 1 for enthalpy integrations). | ||
| T p,onset [°C] | T p,max [°C] | ΔH [J g−1] | T d,onset [°C] | T d,max [°C] | Char [%, 800 °C] | LOI | |
|---|---|---|---|---|---|---|---|
| a Bisphenol-A/aniline benzoxazine (BA-a) with rice straw soda lignin (SL) as published by El Mansouri et al.17 | |||||||
| PL | — | — | — | 292 | 358 | 17.3 | 24.4 |
| BA-aa | 249 | 261 | 277 | 268 | 299 | 25.7 | 27.8 |
| BA-a–SL (7:3)a |
176 | 212 | 303 | 316 | 375 | 40.8 | 33.8 |
| PL-a | 190 | 230 | 117 | 328 | 424 | 47.5 | 36.5 |
| PL-pgl | 154 (156) | 225 (185) | 224 | 329 | 419 | 51.9 | 38.3 |
| oC-pgl | 185 (165) | 215 (188) | 1130 | 326 | 401 | 28.3 | 28.8 |
| pC-pgl | 175 (161) | 210 (184) | 1192 | 329 | 401 | 31.5 | 30.1 |
In the polymerization of propargyl benzoxazines (Fig. 10), two exothermal signals were recorded. The smaller exotherm occurring at a slightly lower temperature was attributed to the polymerization of propargyl groups while the larger exotherm corresponded to the polymerization of benzoxazines. All three propargyl benzoxazines showed similar polymerization temperatures (Table 1), with PL-pgl having lower onset (i.e. for polymerization of both, propargyl and benzoxazine groups) and higher maximum temperature (i.e. for polymerization of benzoxazine groups).
In quantification of DSC signals, lignin-based benzoxazines showed significantly lower enthalpies of polymerization (ΔH in Table 1) compared to the two model benzoxazine compounds despite having the same end-capping moieties, an indication of the lower benzoxazine density in the former. This was attributed to the inherent oligomeric nature of lignin, which was less saturated with phenols qualified for the formation of benzoxazine rings, i.e. in comparison to the phenols used for the synthesis of the model benzoxazine compounds. In addition, the higher enthalpy of polymerization for PL-pgl compared to PL-a was due to the cyclotrimerization of ethynyl groups.
The characterization of thermal stability by TGA (Fig. 11) revealed that onset temperatures of the decomposition reactions were largely comparable (Table 1). Only the lignin-free reference BA-a and the phenolated lignin PL decomposed earlier. A similar trend was observed for the maximum temperatures of decomposition.
 | ||
| Fig. 11 TGA thermograms of phenolated lignin PL, resulting benzoxazines PL-a and PL-pgl as well as model compounds pC-pgl and oC-pgl. | ||
In a review on lignin thermal degradation properties, a multitude of lignins showed maximum decomposition temperatures mostly around 360 °C (ranging from about 240 °C to 390 °C),50 which are in good agreement with PL. Among all samples, benzoxazines synthesized from lignin had the highest temperature maxima at 419 °C and 424 °C and thus the highest thermal stability. For lignin-based epoxy resins, a comparable high decomposition temperature of 416 °C was also found in literature.51
As another measure for thermal stability, the amount of char formation was studied at 800 °C (Table 1). For both lignin-based benzoxazines, the char formation was approximately three times higher than for phenolated lignin alone; and approximately two times higher than those of the two model compounds. With the potential formation of new benzene rings from cyclotrimerization, the char yield of PL-pgl was higher than PL-a by about 9%. The limiting oxygen index (LOI), a parameter used to evaluate the degree of flammability of materials, was calculated using the van Krevelen and Hoftyzer equation with LOI = 17.5 + 0.4 × (char yield).52,53 According to the class scheme by Nelson,54 PL and BA-a were both slow burning materials (LOI ≈ 21 to 28). Although the suggested criteria for self-extinguishing LOI value vary depending on the literature, the material with LOI values greater than 28 were proposed to be self-extinguishing by Fenimore55 and Nelson.54 Under this criterion of self-extinguishing, both lignin-based benzoxazines in Table 1 are self-extinguishing and better than those compared. Recently, an enhanced LOI value was similarly reported when adding increasing amounts of Kraft and organosolv lignin to epoxy resins. The authors obtained a maximum LOI of 32.7, which was exceeded by the lignin–benzoxazines presented here.51
The propargyl group is known to impart good thermal stability. Both pC-pgl and oC-pgl possess higher propargyl concentration per unit molecular weight than PL-pgl. Furthermore, PL-a has no propargyl group in its monomer structure. Despite these facts, PL-a and PL-pgl showed better thermal stability. However, it should be pointed out that the two model compounds used are based on monofunctional phenolic compounds. It is well known in the literature that monofunctional benzoxazines do not polymerize into large molecular weight polymers and form small oligomers of typical molecular weight of 500–2000 g mol−1.8 On the other hand, the lignin-based benzoxazines has already larger molecular size prior to the polymerization reaction. Therefore, it can be safely concluded that this significant improvement in the char yield is the synergistic combination of benzoxazine group as part of the lignin molecule, rather than the intrinsic thermal stability of the propargyl-related groups formed.
4 Conclusions
For the first time, the syntheses of renewable aniline- and propargylamine-benzoxazines were developed solely using organosolv lignin in its phenolated form as aromatic hydroxyl group feedstock. The structural characterization by 31P, 1H, 13C and HSQC NMR spectroscopy in combination with elemental and FT-IR analyses showed that p-hydroxyphenyl and guaiacyl units were successfully converted to benzoxazine units. The density of benzoxazine units formed was similar for both amines and was determined to be as high as 3.2 mmol g−1. The characterization of the polymerization process by isothermal in situ FT-IR studies showed that both lignin-based benzoxazines completed polymerization in an hour or less at 180 °C. Further thermal analysis by DSC and TGA revealed comparable behavior of lignin-based benzoxazines and model compounds with regard to polymerization and decomposition temperatures. The thermal stability, given by the amount of char formed during decomposition, was tremendously increased for both lignin-based benzoxazines, being three times higher than the phenolated lignin and almost two times higher than the model compounds. The propargyl-based lignin-based benzoxazine formed 9% more char than its aniline-based counterpart due to the additional benzene rings formed from cyclotrimerization of terminal ethynyl groups. Moreover, the limited oxygen indices of lignin-based benzoxazines (i.e. >28) led to its classification as self-extinguishing materials, pointing towards incorporation in fire retardant applications.Acknowledgements
One of the authors (G. J. A.) is indebted to the scholarship offered by Engineering Research and Development for Technology (ERDT) under the auspices of the Department of Science and Technology (DOST) of the Philippine government. The research was also funded by the Federal Ministry of Food and Agriculture (BMEL), and supported by the Fachagentur Nachwachsende Rohstoffe e. V. (FNR projects: Lignocellulose-Bioraffinerie II, no. 22019009 and ProLignin, no. 22020811). The authors gratefully acknowledge Rosanna Buchholz (Thünen Institute of Wood Research, TI), Andreas Schreiber (University of Hamburg), Sascha Lebioda (TI), and Christiane Riegert (TI) for their experimental and technical support.Notes and references
- W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259–276 CrossRef CAS.
- S. Canas, M. C. Leandro, M. I. Spranger and A. P. Belchior, Holzforschung, 2000, 54, 255–261 CrossRef CAS.
- O. Anjos, C. Carmona, I. Caldeira and S. Canas, BioResources, 2013, 8, 4484–4496 CrossRef.
- E. Adler, Wood Sci. Technol., 1977, 11, 169–218 CrossRef CAS.
- Y. Lai and K. Sarkanen, in Lignins: Occurrence, formation, structure and reactions, ed. K. Sarkanen and C. Ludwig, John Wiley & Sons, Inc., New York, 1971, pp. 165–240 Search PubMed.
- P. Whiting and D. A. I. Goring, Sven. Papperstidn., 1981, 84, 120–122 Search PubMed.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef CAS.
- H. Ishida, in Handbook of benzoxazine resins, ed. H. Ishida and T. Agag, Elsevier B.V., Amsterdam, 2011, pp. 3–81 Search PubMed.
- C. Amen-Chen, H. Pakdel and C. Roy, Bioresour. Technol., 2001, 79, 277–299 CrossRef CAS PubMed.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrel, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttämaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- K. Chiou and H. Ishida, Curr. Org. Chem., 2013, 17, 913–925 CrossRef CAS.
- N. K. Sini, J. Bijwe and I. K. Varma, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 7–11 CrossRef CAS.
- E. Calò, A. Maffezzoli, G. Mele, F. Martina, S. E. Mazzetto, A. Tarzia and C. Stifani, Green Chem., 2007, 9, 754–759 RSC.
- H. Kimura, Y. Murata, A. Matsumoto, K. Hasegawa, K. Ohtsuka and A. Fukuda, J. Appl. Polym. Sci., 1999, 74, 2266–2273 CrossRef CAS.
- H. Xu, Z. Lu and G. Zhang, RSC Adv., 2012, 2, 2768–2772 RSC.
- J. Podschun, B. Saake and R. Lehnen, Eur. Polym. J., 2015, 67, 1–11 CrossRef CAS.
- N.-E. El Mansouri, Q. Yuan and F. Huang, J. Appl. Polym. Sci., 2012, 125, 1773–1781 CrossRef.
- H. M. E. Haque, Z. Islam, T. Kawauchi and T. Takeichi, Appl. Mech. Mater., 2012, 217–219, 571–577 CrossRef CAS.
- T. Ougi, et al., WO 2013031039 A1, 2013.
- M. Comí, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4894–4903 CrossRef.
- J. Podschun, A. Stücker, B. Saake and R. Lehnen, ACS Sustainable Chem. Eng., 2015, 3, 2526–2532 CrossRef CAS.
- H. Ishida and H. Y. Low, Macromolecules, 1997, 30, 1099–1106 CrossRef CAS.
- H. Ishida and D. Allen, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 1019–1030 CrossRef CAS.
- M. Kanchanasopa, N. Yanumet, K. Hemvichian and H. Ishida, Polym. Polym. Compos., 2001, 9, 367–375 CAS.
- S. W. Kuo, Y. C. Wu, C. F. Wang and K. U. Jeong, J. Phys. Chem. C, 2009, 113, 20666–20673 CrossRef CAS.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- M. Zawadzki and A. Ragauskas, Holzforschung, 2001, 55, 283–285 CrossRef CAS.
- E. A. Capanema, M. Y. Balakshin and J. F. Kadla, J. Agric. Food Chem., 2004, 52, 1850–1860 CrossRef CAS PubMed.
- F. Tran, C. S. Lancefield, P. Kamer, T. Lebl and N. Westwood, Green Chem., 2015, 17, 244–249 RSC.
- A. Nagai, Y. Kmel, Y. Wang, M. Omura, A. Sudo, H. Nishida, E. Kawamoto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2316–2325 CrossRef CAS.
- G. M. Irvine, Tappi J., 1984, 67, 118–121 CAS.
- W. Fiddler, W. E. Parker, A. E. Wasserman and R. C. Doerr, J. Agric. Food Chem., 1967, 15, 757–761 CrossRef CAS.
- J. Rodrigues, J. Graça and H. Pereira, J. Anal. Appl. Pyrolysis, 2001, 58–59, 481–489 CrossRef CAS.
- R. Alén, E. Kuoppala and P. Oesch, J. Anal. Appl. Pyrolysis, 1996, 36, 137–148 CrossRef.
- J. C. del Río, A. Gutiérrez, J. Romero, M. J. Martínez and A. T. Martínez, J. Anal. Appl. Pyrolysis, 2001, 58–59, 425–439 CrossRef.
- A. I. Afifi, J. P. Hindermann, E. Chornet and R. P. Overend, Fuel, 1989, 68, 498–504 CrossRef CAS.
- M. J. Antal Jr, Ind. Eng. Chem. Prod. Res. Dev., 1983, 22, 366–375 CrossRef.
- D. W. van Krevelen, in Properties of polymers; Their correlation with chemical structure; Their numerical estimation and prediction from additive group contributions, ed. D. W. van Krevelen and K. te Nijenhuis, Elsevier B.V., Amsterdam, 4th edn, 2009, pp. 847–873 Search PubMed.
- H. Ishida and J. Liu, in Handbook of Benzoxazine Resins, ed. H. Ishida and T. Agag, Elsevier, Amsterdam, 2011, pp. 85–102 Search PubMed.
- J. M. García, G. O. Jones, K. Virwani, B. D. Mccloskey, D. J. Boday, G. M. Huurne, H. W. Horn, D. J. Coady, A. M. Bintaleb, A. M. S. Alabdulrahman, F. Alsewailem, H. A. A. Almegren and J. L. Hedrick, Science, 2014, 344, 732–735 CrossRef PubMed.
- Z. Brunovska, J. P. Liu and H. Ishida, Macromol. Chem. Phys., 1999, 200, 1745–1752 CrossRef CAS.
- A. E. Wroblewski, C. Lensink, R. Markuszewski and J. G. Verkade, Energy Fuels, 1988, 2, 765–774 CrossRef CAS.
- H. J. Kim, Z. Brunovska and H. Ishida, Polymer, 1999, 40, 6565–6573 CrossRef CAS.
- K. D. Demir, B. Kiskan and Y. Yagci, Macromolecules, 2011, 44, 1801–1807 CrossRef.
- National Institute of Advanced Industrial Science and Technology, Spectral database of organic compounds, SDBS, http://sdbs.db.aist.go.jp.
- K. Lundquist, in 11th International Symposium on Wood and Pulping Chemistry, Nice, France, 2001, pp. 1–4 Search PubMed.
- M. J. Lambregts, E. J. Munson, A. A. Kheir and J. F. Haw, J. Am. Chem. Soc., 1992, 114, 6875–6879 CrossRef CAS.
- P. Pichat, J. Vedrine, P. Gallezot and B. Imelik, J. Catal., 1974, 32, 190–203 CrossRef CAS.
- D. A. I. Goring, in Lignins: occurrence, formation, structure and reactions, ed. K. V. Sarkanen and C. H. Ludwig, Wiley-Interscience, New York, 1971, pp. 695–865 Search PubMed.
- M. Brebu and C. Vasile, Cellul. Chem. Technol., 2010, 44, 353–363 CAS.
- F. Ferdosiana, Z. Yuana, M. Anderson and C. Charles Xu, J. Anal. Appl. Pyrolysis, 2016, 119, 124–132 CrossRef.
- D. van Krevelen and P. Hoftyzer, Properties of Polymers, Elsevier, New York, 2nd edn, 1976 Search PubMed.
- D. W. van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- M. I. Nelson, Combust. Theory Modell., 2001, 5, 59–83 CrossRef CAS.
- C. P. Fenimore, in Flame-Retardant Polymeric Materials, ed. M. Lewin, S. M. Atlas and E. M. Pearce, Plenum Press, New York, 1975, pp. 371–397 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |