
Insights into choline chloride–phenylacetic acid deep eutectic solvent for CO2 absorption†
Tausif Altamasha,
Mert Atilhan*a,
Amal Aliyana,
Ruh Ullaha,
Gregorio García b and
Santiago Aparicio*b
b and
Santiago Aparicio*b
aDepartment of Chemical Engineering, Qatar University, Doha, Qatar. E-mail: mert.atilhan@qu.edu.qa
bDepartment of Chemistry, University of Burgos, Burgos, Spain. E-mail: sapar@ubu.es
First published on 7th November 2016
Abstract
The properties of choline chloride plus phenylacetic acid deep eutectic solvents in neat liquid state and upon absorption of CO2 are analyzed using a theoretical approach combining quantum chemistry using Density Functional Theory and classic molecular dynamics methods. This study investigates the physicochemical properties, structuring, dynamics and interfacial behavior of the selected deep eutectic solvent from the nano-size point of view to infer its viability for effective CO2 capture. DFT results provided information on the mechanism of short-range interactions between CO2 and the studied DES, showing a better performance than previously studied DES. The mechanism of CO2 capture is analyzed considering model flue gas, showing a two-stage process with water, CO2 and N2 molecules developing adsorbed layers at the interface but in different regions. Water adsorbed layers would delay the migration of CO2 molecules toward bulk liquid regions, which should be considered for developing large-scale applications.
1. Introduction
The increasing atmospheric CO2 concentration,1 which is caused in a large percentage by emissions coming from electricity production using fossil fuels,2 has led to the need of developing carbon capture technologies.3,4 The use of amine-based sorbents,5 which is a well-known technology applied for decades in the gas industry, would be the first possibility, but the large number of technological problems (corrosion, sorbent degradation, high energy penalty, etc.)6–8 shows the need for new methods. For this purpose, many different types of new sorbents have been studied in academia,9–14 although the industrial scale application of most of them has not been tested.15 Ionic liquids (IL) have been considered one of the most promising options for developing carbon capture methods,16–20 mainly considering the possibility of designing specific ionic combinations with suitable properties. The task-specific characteristic of IL is a clear advantage or IL over other possible sorbents, even more remarkable considering the large amount of anion–cation combinations leading to room temperature IL. Nevertheless, developing the most suitable IL for carbon capture purposes requires a deep knowledge of structure–property relationships and of IL properties at the molecular level. Likewise, some properties of some IL, such as high viscosity, poor biodegradability or large cost, has been showed,21–26 which would difficult the development of carbon capture technologies. These problems have led to some authors to show doubts about the suitability of IL27 and the need of searching alternative or complementary technologies. Therefore, although the large number of ionic combinations is a flexible library for finding the most suitable ionic combinations and thus circumventing the problems,28 the exploration of related alternatives is advisable.Deep Eutectic Solvents (DES) have been recently proposed as possible platforms for developing carbon capture sorbents.29–32 DES are mixtures with of two, or more, compounds with melting temperature lower than either of the individual compounds.33–35 These eutectic mixtures are formed by combining Lewis or Brønsted acids and bases which can contain different types of ionic species.35 The depression of the melting point upon mixing of the DES compounds are produced by the hydrogen bonding interaction between the DES compounds, and thus, the mixing of an hydrogen bond acceptor (HBA), frequently a quaternary ammonium salt, and an hydrogen bond donor (HBD), is the common approach for developing DES.
The main DES properties and their suitability for carbon capture purposes have been recently reviewed,30 and although the number of studies is still limited the interest on these systems has increased recently. The main advantages of DES rise from the large number of compounds leading to DES, which produce a large library for selecting the most suitable DES according to their physicochemical properties and affinity for CO2. Likewise, DES synthesis carried out with 100% atom economy,36 the null toxicity and full biodegradability37,38 of most of used HBA and HBD, the possibility of developing DES based on fully natural molecules,39 and their low cost, are remarkable advantages for DES.
The developing of DES-based technologies require to find the most suitable HBA–HBD combinations, leading to the best physicochemical properties (e.g. low viscosities, large thermal stability) and high affinity for CO2 molecules. For this purpose, understanding the microscopic behavior of DES, its relationship with macroscopic physicochemical properties, and the mechanism of interaction between CO2 and DES-molecules is of pivotal importance. This information can be obtained using computational chemistry methods such as Density Functional Methods (DFT)40,41 and classic molecular dynamics (MD).42–44 The use of computational chemistry for DES have led to information on the structuring, dynamics and CO2-capturing mechanism for selected systems, although its systematic application to understand DES properties is still in its infancy.40–44 For this purpose, the properties of DES based on choline chloride as HBA (CHCl) and phenylacetic acid (PhOAc) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mole ratios have been studied in this work using both DFT and MD approaches. Our group has published previous studies in which the properties of DES based on natural products, such as levulinic acid, were analyzed.42 The objective of the present work is to extend systematically the results of previous studies to DES based on PhOAc as HBD and to infer the changes in the microscopic properties of the DES in relationship with CO2 capture. The selection of PhOAc was done as a model of aromatic carboxylic acids as a platform for DES development. The presence of the bulky phenyl ring in PhOAc should contribute to increase the available free volume in the fluid, which could favor CO2 solubility combined with the presence of the carboxylic acid group. The purpose of this work is to characterize this type of material as part of the research on the suitability of DES for carbon capturing in order to find the most suitable molecular combinations and their relationship with molecular features and nano-sized liquid properties.
:2 mole ratios have been studied in this work using both DFT and MD approaches. Our group has published previous studies in which the properties of DES based on natural products, such as levulinic acid, were analyzed.42 The objective of the present work is to extend systematically the results of previous studies to DES based on PhOAc as HBD and to infer the changes in the microscopic properties of the DES in relationship with CO2 capture. The selection of PhOAc was done as a model of aromatic carboxylic acids as a platform for DES development. The presence of the bulky phenyl ring in PhOAc should contribute to increase the available free volume in the fluid, which could favor CO2 solubility combined with the presence of the carboxylic acid group. The purpose of this work is to characterize this type of material as part of the research on the suitability of DES for carbon capturing in order to find the most suitable molecular combinations and their relationship with molecular features and nano-sized liquid properties.
2. Methods
2.1 DFT study
In concordance with our previous works dealing with DFT studies on DES solvents for CO2 mitigation,40–44 all the reported DFT results were obtained out by using B3LYP45–47 coupled with dispersion corrections according to Grimme's scheme (B3LYP-D2),48 in combination with 6-31+G** basis set. In this sense, B3LYP has proven its performance over a wide range of systems,49 while dispersion corrections are adequate for systems with dispersive interactions such as hydrogen bonds.50 DFT studies were carried out for CHCl_PhOAC_1_2 at 1:2 molar ratios.
Geometry optimizations for molecular clusters composed of 1 CHCl ionic pair + 2 PhOAc molecules and for the same system + 1 CO2 molecule at different positions were developed with the Berny algorithm using GEDIIS.51 The optimized structures were checked through their vibrational frequencies, discarding the presence of negative frequencies. Computed energies were corrected (to avoid basis set superposition error) according to counterpoise procedure.52 Atomic charges were computed to fit the electrostatic potential according to the ChelpG scheme.53 All calculations were carried out with Gaussian 09 (Revision D.01) package.54 Interaction energies (ΔE) for the studied processes related with binding energy for [CH][Cl] salt (ΔEIP), DES formation (ΔEDES) and CO2 capture by the selected solvent (ΔEDES–CO2) were defined as:
| ΔEIP = EIP − (ECH + ECl) | (1) |
| ΔEDES = EDES − (EIP + 2EPhOAc) | (2) |
| ΔEDES·CO2 = EDES·CO2 − (EDES + ECO2) | (3) |
2.2 MD simulations
The first objective of MD simulations was to analyze the properties of pure CHCl_PhOAc_1_2 (CHCl_PhOAc of 1:2 ratio) and for the mixtures of this DES with CO2 molecules. For the characterization of DES properties in absence of CO2 molecules (pure DES), simulation boxes containing 250 [CH][Cl] ion pairs plus 500 PhOAc molecules (1:2 mole ration) were used and NPT simulations in the 298 to 358 K temperature range and 0.1 MPa were carried out. Regarding the study of mixed CHCl_PhOAc_1_2 + CO2, systems were built according to the experimental solubility data obtained in our laboratories (Table S1, ESI†) and NPT simulations at 298 K and the corresponding pressures were carried out. Initial cubic boxes were built using the Packmol program,55 from 5 ns equilibration runs were done (with equilibration assured by constancy of total potential energy) followed by 20 ns runs for production purposes.
The second objective of MD studies was to characterize the interfacial properties of CHCl_PhOAc_1_2 in contact with vacuum layers and with acid gas phases. For this purpose, previously equilibrated simulation boxes (with the same amount of molecules used for the study of pure DES properties) were put in contact with a vacuum layer (200 Å long in the z-direction), with a gas phase containing 500 CO2 molecules (in a box 190 Å long in the z-direction, corresponding to CO2 vapor density equal to 0.066 g cm−3 equal to that experimentally obtained for vapor CO2 at 3 MPa and 298 K),56 or with a gas phase emulating the typical composition of a flue gas, which is the mixture of gases being emitted from fossil-fueled power plants, (50CO2 + 375N2 + 50H2O + 25O2 molecules, in a box 190 Å long in the z-direction). These simulations for the study of interface behavior were carried out in the NVT ensemble at 298 K for CO2 and flue gas interfaces, and at 298, 318, 338 and 358 K for vacuum interfaces.
MDynaMix v.5.2 software was used to carry out all the simulations reported in this work.57 Simulations in the NVT and NPT ensembles were carried out with pressure and temperature controlled with the Nose–Hoover method. Ewald method58 was applied for handling coulombic interactions. The equations of motion were solved using the Tuckerman–Berne double time step algorithm (1 and 0.1 fs, for long and short time steps, respectively).59 Lennard-Jones cross terms were calculated using Lorentz–Berthelot mixing rules.
The forcefield parameterization used along MD simulations is reported in Table S2 (ESI†). According to our previous studies for other types of DES,43 the atomic charges develop a pivotal role for DES MD simulations, and thus, they were obtained from DFT calculations of 1 [CH][Cl] + 2 PhOAc clusters optimized at B3LYP-D2/6-31+G** theoretical level using ChelpG charges.53 In agreement with previous results for other DES,42 the 1:2 stoichiometry of the studied DES leads to two types of PhOAc molecules with slightly different atomic charges because of different interactions with the salt (Table S2, ESI†). Likewise, used total charges are +0.6941 for [CH]+, −0.6707 for Cl−, and +0.0261 and −0.0497 for the two different types of PhOAc molecules. Lennard-Jones parameters for [CH]+ and Cl− were obtained from a previous work,44 whereas those for PhOAc were obtained from SwissParam.60 Forcefield parameters for gas molecules were obtained from a previous work.61
3. Results and discussion
3.1 DFT results
Prior to analyzing CO2 capture using DFT by CHCl_PhOAc_1_2 DES, this paragraph briefly discusses the main features for choline chloride ionic compounds and CHCl_PhOAc_1_2 deep eutectic solvent (Fig. S1, ESI†). As previously reported,41 the ionic compound yields a high interaction energy (ΔEIP = −518.81 kJ × mol−1) due to the coulombic attraction between ions, with an intermolecular charge transfer equal to 0.161e−. Although, the chloride anion leads to four intermolecular hydrogen bonds with the cation, the main interaction is via the OH group (with a bond length equal to 2.084 Å). Regarding to CHCL_PhOAc_1_2, its optimized structure is mainly characterized by intermolecular hydrogen bonding between the chloride ion and both acetic motifs, with bond lengths of around 2.146 Å. Likewise, both phenyl acetic molecules also establish some hydrogen bonds with the choline cation. The formation energy for CHCl_PhOAc_1_2 was ΔEDES = −155.86 kJ × mol−1. In addition, some negative charge is transferred from the anion up to one phenyl acetic molecule (0.050e−), while the other hydrogen bond doctor becomes to be slightly positive charged (0.026e−).Fig. 1 shows the optimized structures for CHCl_PhOAc_1_2⋯CO2. For a first approximation, CO2 capture at the molecular level would be related with the strength of the interactions between the solvent (CHCl_PhOAc_1_2) and the gas molecule, which has been assessed through interaction energies defined above (eqn (3)). Aimed at obtaining the most relevant information on the potential energy surface for the interaction between CHCl_PhOAc_1_2 and CO2 molecule, several starting points for CHCl_PhOAc_1_2 and one CO2 molecule in different relative positions were tested, selecting those molecular arrangements with lower total energies. As seen in Fig. 1, nine arrangements were found for the interaction between the DES and the gas molecule. This figure also reports interaction energies related with CO2 capture, which are ranged between 53.65 kJ × mol−1 (structure 9) and 22.78 kJ × mol−1 (structure 6). For structure 9, the CO2 molecule is mainly linked (concretely the central carbon atom) to the Cl atom with a bond length equal to 3.342 Å and to the O atom (from COOH motif) with a bond length equal to 2.902 Å. Structures 5 (ΔEDES·CO2 = 42.87 kJ × mol−1) and 7 (ΔEDES·CO2 = 39.94 kJ × mol−1) yields a similar interaction mechanism between DES motif and CO2 molecule. For structures 8 (ΔEDES·CO2 = 42.80 kJ × mol−1), 2 (ΔEDES·CO2 = 36.08 kJ × mol−1), 3 (ΔEDES·CO2 = 31.92 kJ × mol−1) and 4 (ΔEDES·CO2 = 31.92 kJ × mol−1), CO2 molecule is placed in the vicinity of an oxygen atom (from COOH motif), with a bond length ≈ 2.834 Å. Considering that the hydrogen atom of the hydroxyl group in PhOAc is interacting with chlorine anion, the interaction of CO2 molecules for structures 2, 3, 4 and 8 is in agreement with the available DFT studies on the interaction of CO2 with organic acids.62 Energy differences could be related with the strength of C(CO2)⋯O(COOH) interactions as well as the presence of some weak hydrogen bond between CO2 molecule (through H atoms) and methyl H (from choline cation). For structures 1 (ΔEDES·CO2 = 43.70 kJ × mol−1) and 6 (ΔEDES·CO2 = 22.78 kJ × mol−1), the main interaction is related with and CO2–Cl intermolecular bond. Nonetheless, the absence of additional H-bond between CO2 molecules and choline cation (there is only a weak H-bond between CO2 and phenyl motif) in structure 6 leads to the lowest |ΔEDES·CO2| values. The interaction energies obtained for CHCl_PhOAc_1_2 from DFT would agree with a better performance for CO2 capture in comparison with other deep eutectic solvents previously reported by our group, such as the one based on levulinic acid as hydrogen bond donor.42
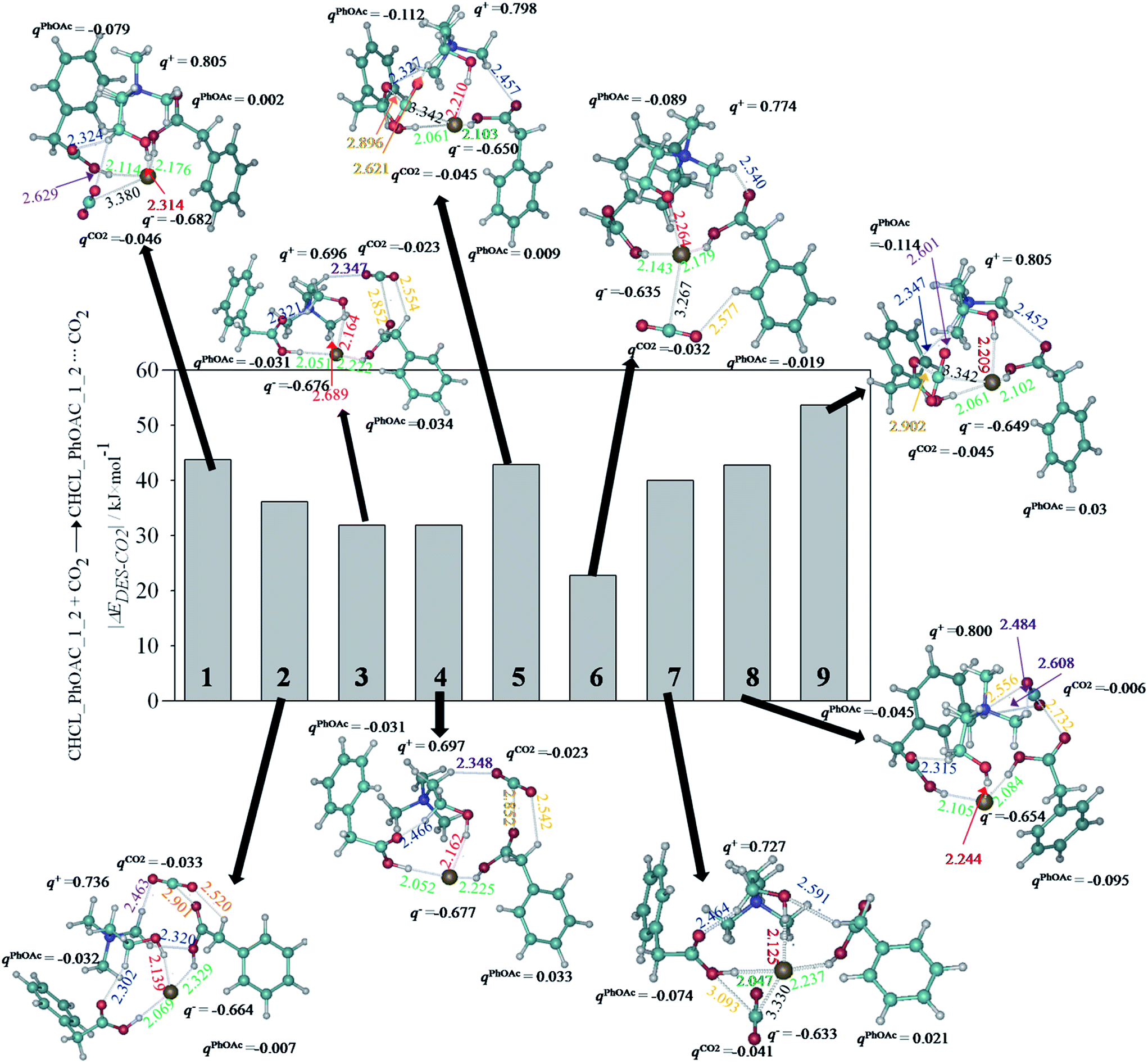 | ||
| Fig. 1 Optimized structures for CHCl_PhOAc_1_2⋯CO2 system (nine different arrangements were found) at B3LYP-D2/6-31+G** level, along the main structural parameters related with intermolecular interactions. Computed charges over choline (q+), chloride (q−), phenyl acetic (qPhOAc) and CO2 (qCO2) motifs, as well as the binding energy (ΔEDES–CO2) corresponding for CO2 catch by selected DES are also shown. Intermolecular bond lengths are in Å. | ||
3.2 MD results
MD simulations were also carried out for CHCl_PhOAc_1_2. The comparison of thermophysical properties obtained from MD simulations and experimental data are reported in Fig. S2 (ESI†). Regarding predicted density, an excellent agreement is obtained, Fig. S2a (ESI†), with predicted data slightly larger than experimental values but with 1.1% average deviation in the simulated temperature range. Likewise, the evolution of density with temperature is also accurately predicted, which leads to excellent predictions of the thermal expansion coefficient, Fig. S2b (ESI†). Viscosity predictions are reported in Fig. S2c (ESI†) showing a reasonable agreement between experiments and simulations, especially at low temperatures, which shows how the model used for MD studies is able to give a reasonable description of the dynamic properties of the studied system. Self-diffusion coefficients, D, reported in Fig. S2d (ESI;† calculated from mean square displacements and Einstein's equation) could not be compared with experimental results but follow the ordering D(PhOAc) > D(Cl−) > D([CH]+), in agreement with previous results for DES containing levulinic acid.42 Nevertheless, the evolution of self-diffusion coefficients with temperature is parallel for the ions and PhOAc, showing highly correlated molecular diffusion because of the hydrogen bonding between ions and PhOAc molecules.The structure of CHCl_PhOAc_1_2 at the nano-sized level is firstly analyzed using the radial distribution functions, RDFs, reported in Fig. 2. Results for center-of-mass RDFs reported in Fig. 2a confirm the strong anion–cation interaction in the DES together with the preferential interaction of PhOAc with chlorine anion. The anion–cation interaction is developed through the hydroxyl group in [CH]+ (O1–H4 sites) and PhOAc interacts with Cl− through the hydroxyl group (O2–H5 sites). No hydrogen bonding between PhOAc and [CH]+ is inferred, whereas hydrogen bonding between neighbor PhOAc molecules is inferred, Fig. 2b. The spatial distribution around [CH]+ reported in Fig. 3a shows anionic concentration around the head hydroxyl group with additional distribution around the methyl groups bonded to nitrogen but discarding hydrogen bonding. In the case of distribution around PhOAc, Fig. 3b, Cl− is placed above the –OH group and neighbor PhOAc molecules in close regions around the –COOH group.
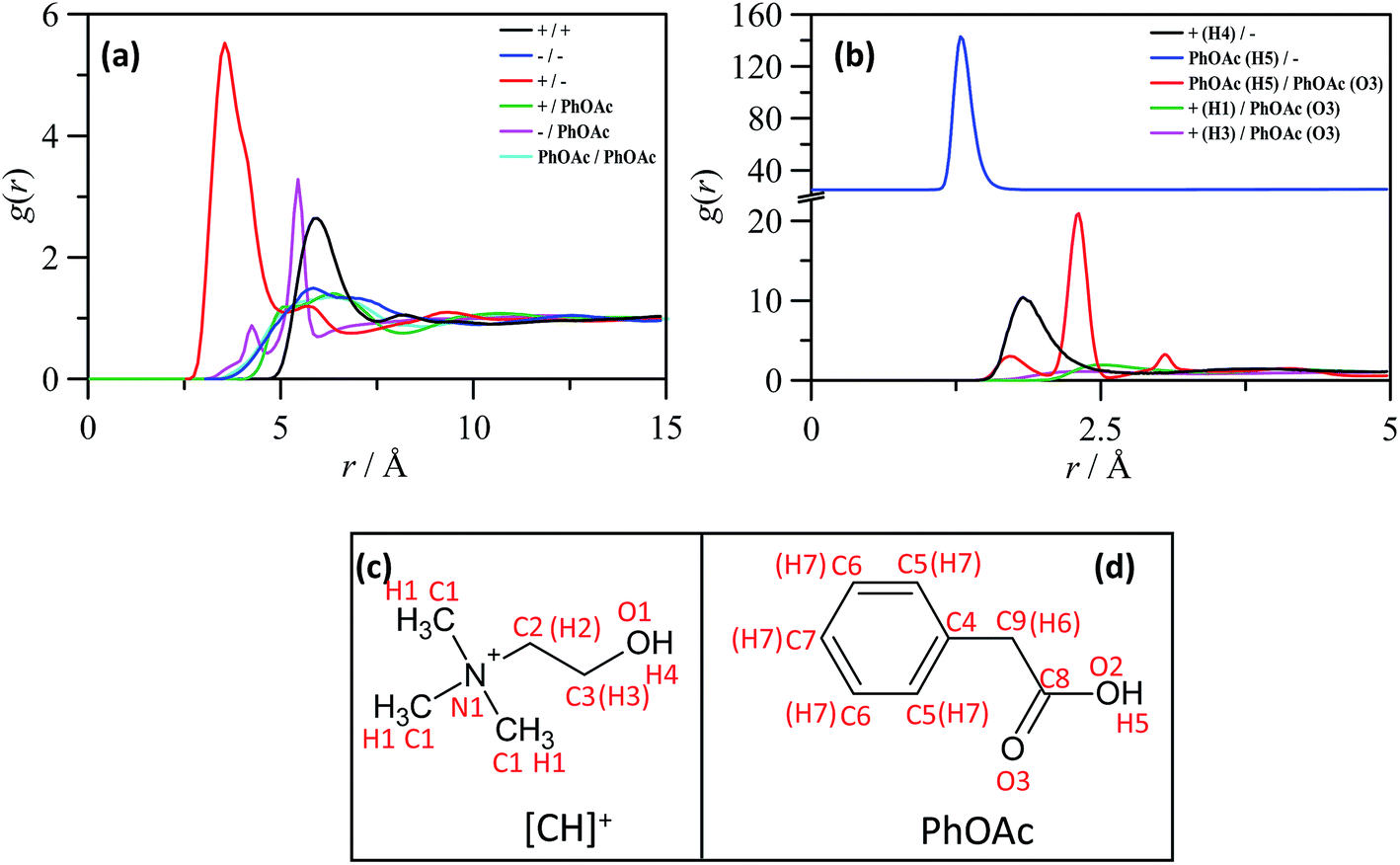 | ||
| Fig. 2 Radial distribution functions, g(r), obtained from molecular dynamics simulations for CHCl_PhOAc_1_2 at 298 K and 1 bar. Panel (a) shows center-of-mass/center-of-mass g(r) and panel (b) site–site g(r). + stands for [CH]+ and − for Cl−. Panels (c) and (d) shows atom labelling used for the analysis of molecular dynamics results. | ||
 | ||
| Fig. 3 Spatial distribution functions for the reported sites around (a) [CH]+ and (b) PhOAc obtained from molecular dynamics simulations for CHCl_PhOAc_1_2 at 298 K and 1 bar. Isodensity values reported for 4-times bulk density. Atom names as in Fig. 2. | ||
The dynamics of solvation shells around each molecule are quantified using the residence times of one molecule around other, as defined in a previous work,42 Fig. S3 (ESI†). These results show that PhOAc molecules stay longer times in the vicinity of Cl− than [CH]+, which confirm the strong affinity of PhOAc for the anion. The residence times of one PhOAc molecule around another neighbor PhOAc molecule are also large confirming the development of self-association between these molecules. This is in agreement with the strength of the intermolecular forces reported in Fig. S4 (ESI†), which show strong anion–cation interactions (as expected from their prevailing coulombic character), but also strong PhOAc–Cl− interactions. Likewise, the average number of hydrogen bonds reported in Fig. 4 confirms the development of hydrogen bonding between PhOAc and Cl−, which is surprisingly even slightly reinforced with increasing temperature and the minor extension of anion–cation and PhOAc–PhOAc interactions.
 | ||
| Fig. 4 Number of hydrogen bonds between the reported sites, nH-bonds, for CHCl_PhOAc_1_2 at 298 K and 1 bar. Values reported per [CH]+ (for H4/Cl) or per PhOAc. Donor–acceptor distance of 3.0 Å and 60° angle were used as cutoffs for hydrogen bond definition. Atom names as in Fig. 12. | ||
The absorption of CO2 molecules should lead to changes in the structure of the studied CHCl_PhOAc_1_2 DES. First, the liquid should show expansion upon gas absorption but this effect is very minor with an expansion of just 12% for xCO2 = 0.48, Fig. 5, which shows that the DES is able to rearrange its structure for fitting the absorbed gas molecules without very remarkable changes in DES liquid structuring. This is confirmed by the intermolecular interaction energies between of CHCl_PhOAc_1_2 molecules reported in Fig. 6a, which suffer very minor changes upon CO2 absorption in the studied range. Moreover, the affinity of PhOAc and [CH]+ molecules for CO2 is very similar, Fig. 6b, but the strength of PhOAc–CO2 interactions is the half of levulinic acid–CO2 interactions as reported in a previous work,20 but CO2 solubility is almost the same both in DES containing PhOAc and levulinic acid (both DES with the same ions and stoichiometry), showing the relevance of other factors such as volume rearrangement upon gas absorption. The arrangement of CO2 molecules in CHCl_PhOAc_1_2 is analyzed from RDFs reported in Fig. 7. RDFs around [CH]+ show that CO2 molecules are preferentially placed around the cation hydroxyl group, Fig. 7a, which leads to a peak in RDFs around Cl−, Fig. 7b. Regarding the distribution around PhOAc, results in Fig. 7c show that CO2 molecules are preferentially placed around the aromatic ring in PhOAc. This is confirmed by the spatial distribution functions reported in Fig. 8. Likewise, results in Fig. S5 (ESI†) show a trend for developing CO2–CO2 clustering with increasing CO2 mole fraction, especially around PhOAc as reported in Fig. 8b. Regarding the dynamics of CO2 molecules, results in Fig. S6 (ESI†) show that in spite of the moderate strength of intermolecular interactions involving CO2 molecules, Fig. 6b, the residence times of CO2 are long enough when compared with other stronger interactions, Fig. S3 (ESI†).
 | ||
| Fig. 5 Percentage of volume change, % Vexp, upon CO2 absorption, according to the criteria by Gallagher et al.,63 for CHCl_PhOAc_1_2 + CO2 mixtures at 298 K as a function of CO2 mole fraction, xCO2. | ||
 | ||
| Fig. 6 Intermolecular interaction energy, Einter, sum of Lennard-Jones and coulombic terms, for CHCl_PhOAc_1_2 + CO2 at 298 K and 1 bar. | ||
 | ||
| Fig. 7 Site–site radial distribution functions, g(r), for CHCl_PhOAc_1_2 + CO2 systems at 298 K and 29.91 bar (xCO2 = 0.478). Atom names as in Fig. 12; CD stands for carbon atoms in CO2. | ||
 | ||
| Fig. 8 Spatial distribution functions of CO2 center-of-mass around (a) [CH]+ and (b) PhOAc for CHCl_PhOAc_1_2 + CO2 at 298 K and 29.91 bar (xCO2 = 0.478). Values reported for 4 times bulk density. | ||
The absorption of CO2 molecules from a gas phase is developed in two stages, first gas molecules are adsorbed at the corresponding interface and second, they diffuse from the interfacial region toward the bulk liquid phase. The first stage, adsorption at the interface, is critical for the development of industrial large scale gas capturing operations because very strong adsorption and long residence times at the interface would hinder the diffusion toward the bulk liquid, which should be considered for process design purposes. Therefore, the nano-sized characteristics of CHCl_PhOAc_1_2 interfaces in contact with a CO2 gas phase and with a model flue gas were also studied using MD. Likewise, the CHCl_PhOAc_1_2–vacuum interface was also studied for comparison purposes. The structure of the interface at vacuum is characterized by regions in contact with the vacuum layer being rich in PhOAc, whereas ions are placed in inner regions close to the Gibbs dividing surface, Fig. 9a. The atomic arrangement at vacuum interface, Fig. 9b and c, show that [CH]+ is placed almost parallel to the interface although slightly skewed with hydroxyl groups pointing to the vacuum layer. PhOAc are slightly skewed regarding to the vacuum interface, with –COOH group placed in inner regions and phenyl ring closer to the vacuum layer in parallel to the interface. The contact of CHCl_PhOAc_1_2 with a CO2 gas layer leads to minor changes in the arrangement of molecules at the interface in comparison with vacuum interface, Fig. 9d, and although a certain rearrangement of [CH]+ and PhOAc is produced in contact upon CO2 adsorption the orientation of molecules is very similar, Fig. 9e and f. The contact of CHCl_PhOAc_1_2 with CO2 gas phase leads to the development of a highly dense adsorbed layer in the first stages of the simulation, Fig. S7 (ESI†). This adsorbed layer follows a complex dynamics, first the intensity of the peaks shows increasing number of adsorbed molecules, then the peaks broadening shows that certain CO2 molecules move toward inner regions, and finally the shifting of the peaks show adsorbed layers moving toward inner regions at the interface, Fig. S8 (ESI†). Results in Fig. 9d showed that the outer layers of CHCl_PhOAc_1_2, in contact with CO2 gas phase, are rich in PhOAc molecules, and thus the adsorption of CO2 molecules is characterized by strong PhOAc–CO2 interactions (especially when the adsorbed layer is fully developed, t > 6 ns), and in minor extension interactions with [CH]+ are developed, Fig. 10.
 | ||
| Fig. 9 Density profiles for (a and d) center of mass of [CH]+, Cl− and PhOAc, and for relevant atoms in (b and e) [CH]+ and (c and f) PhOAc, molecules in CHCl_PhOAc_1_2 + (a–c) vacuum and (d–f) CO2 interfaces systems calculated from molecular dynamics simulations at 298 K. z stands for the coordinate perpendicular to the corresponding interfaces, and zGDS for the coordinate of the Gibbs dividing surface. Profiles obtained as averages in the 4 to 5 ns simulation range. Panels at the center of the figure shows the molecular arrangements at the interfaces and the corresponding angles. | ||
 | ||
| Fig. 10 Intermolecular interaction energy, Eint, between CO2 and molecules in CHCl_PhOAc_1_2 + CO2 interface system as a function of simulation time, calculated from molecular dynamics simulations at 298 K. | ||
For industrial operations involving CO2 capturing, the gas phase from which CO2 has to be adsorbed is a mixture (flue gas) in which the low partial pressure of CO2 hinders its capturing. Therefore, the properties of CHCl_PhOAc_1_2 in contact with a model flue gas were also studied in this work. The density profiles at the flue gas interface reported in Fig. 11a show that water molecules present in the flue gas develops an adsorbed layer in inner regions when compared with CO2 adsorbed layer. The water adsorbed layer stays in regions close to Cl− whereas CO2 molecules stay close to PhOAc molecules. Therefore, although the adsorption of water molecules does not hinder the development of a CO2 adsorbed layer, because they occupy different regions at the interface, the water layer should difficult the diffusion of CO2 molecules from the interface toward bulk liquid regions. Moreover, an additional adsorbed layer of N2 molecules is developed at outer regions, and thus three consecutive layers are developed at the interface being occupied by water, CO2 and N2 molecules, Fig. 11b. Likewise, the adsorption mechanism from flue gas is characterized by a moderate affinity toward CO2 molecules paired with very strong water–Cl− interactions and non-negligible N2–PhOAc interactions, Fig. S9 (ESI†).
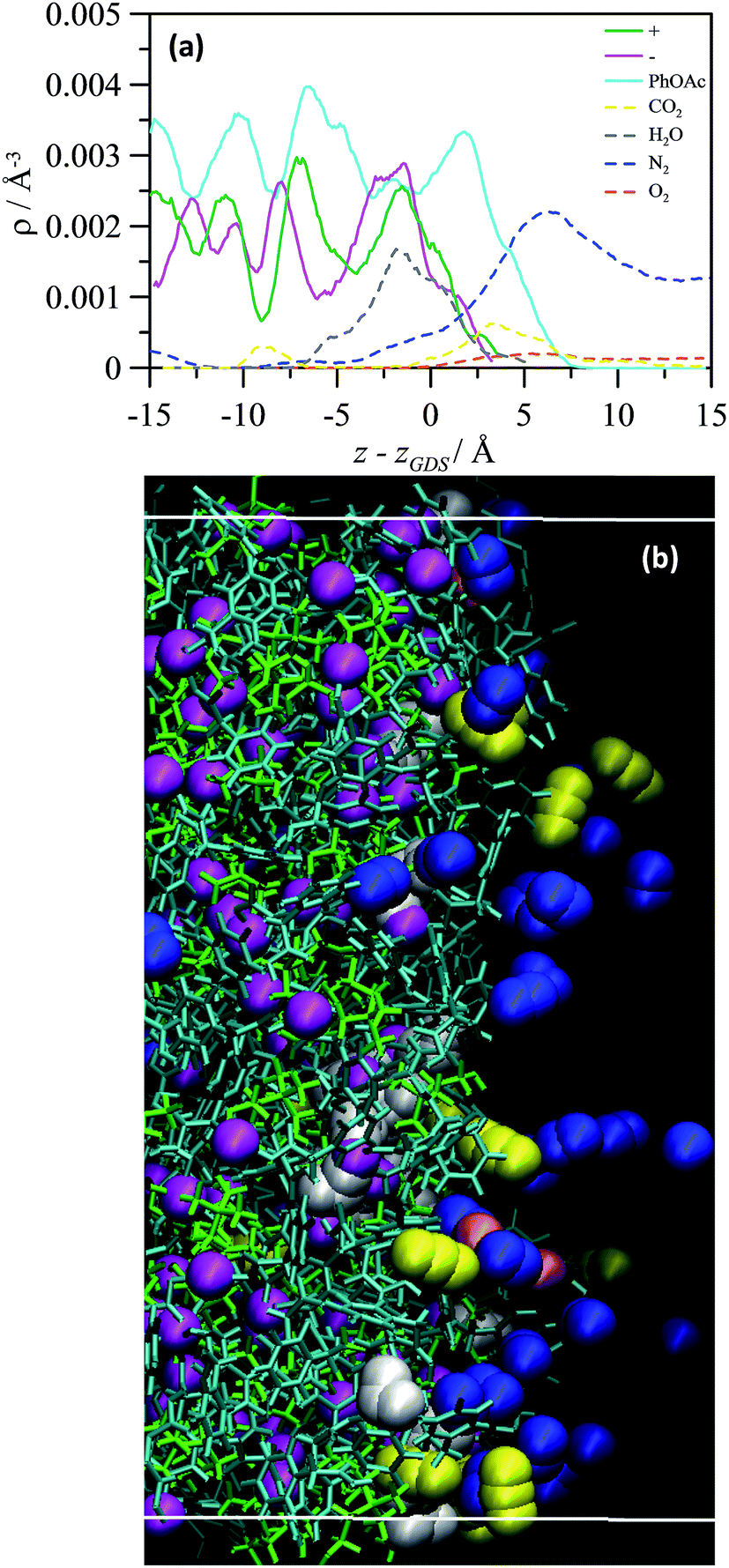 | ||
| Fig. 11 (a) Density profiles for center of mass of [CH]+, Cl−, PhOAc and gas molecules in CHCl_PhOAc_1_2 + flue gas interface system calculated from molecular dynamics simulations at 298 K. z stands for the coordinate perpendicular to the corresponding interfaces, and zGDS for the coordinate of the Gibbs dividing surface. Values calculated in the 0.5 to 1.0 ns simulation time. (b) Snapshot for the same system. | ||
4. Conclusions
The reported theoretical study on choline chloride plus phenylacetic acid led to a detailed characterization of the fluids and the changes upon CO2 capturing. Density Functional Theory results showed large interaction between the components of the deep eutectic solvent and CO2 molecules. The model used for molecular dynamics simulations led to excellent prediction of the most relevant physicochemical properties of the fluid, and thus capturing the physics of the studied solvent. The process of CO2 absorption is characterized by a very minor volume expansion, confirming the ability of the fluid to rearrange its structuring without weakening HBA–HBD intermolecular interactions. The CO2 capturing mechanism, which was studied using a model flue gas, is characterized by a two steps process in which the development of water, CO2 and nitrogen layers is the main step. The very fast development of water adsorbed layers in inner regions of the solvent–gas interface is inferred, which would slow the CO2 diffusion toward bulk liquid regions in the solvent, and should be considered in the design of CO2 capturing operations using the studied deep eutectic solvent.Acknowledgements
This work was made possible by NPRP grant # 6-330-2-140 from the Qatar National Research Fund (a member of Qatar Foundation) and by Ministerio de Economía y Competitividad (Spain, project CTQ2013-40476-R). We also acknowledge The Foundation of Supercomputing Center of Castile and León (FCSCL, Spain) for providing supercomputing facilities. The statements made herein are solely the responsibility of the authors.References
- J. H. Butler and S. A. Montzka, The NOAA annual greenhouse gas index (AGGI), NOAA Earth System Research Laboratory, 2016, updated spring 2016, http://www.esrl.noaa.gov/gmd/ccgg/aggi.html.
- CO2 emissions from fossil fuel combustión. Highlights, International Energy Agency, Paris, 2015 Search PubMed.
- B. Sreenivasulu, D. V. Gayatri, I. Sreedhar and K. V. Raghavan, Renewable Sustainable Energy Rev., 2015, 41, 1324–1350 CrossRef CAS
.
- K. A. Mumford, Y. Wu, K. H. Smith and G. W. Stevens, Front. Chem. Sci. Eng., 2015, 9, 125–141 CrossRef CAS
- Z. Liang, W. Rongwong, H. Liu, K. Fu, H. Gao, F. Cao, R. Zhang, T. Sema, A. Henni, K. Sumon, D. Nath, D. Gelowitz, W. Srisang, C. Saiwari, A. Benamor, M. Al-Marri, H. Shi, T. Supap, C. Chan, Q. Zhou, M. Abu-Zahra, M. Wilson, W. Olson, R. Idem and P. Tontiwachuthikul, Int. J. Greenhouse Gas Control, 2015, 40, 26–54 CrossRef CAS
- J. Kittel and S. Gonzales, Oil Gas Sci Technol – Revue d'IFP Energies nouvelles, 2014, 69, 915–929 CrossRef
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CAS
- P. Luis, Desalination, 2016, 380, 93–99 CrossRef CAS
- N. Hedin, L. Chen and A. Laaksonen, Nanoscale, 2010, 2, 1819–1841 RSC
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 CAS
- R. Ben-Mnasour, M. A. Habib, O. E. Bamidel, M. Basha, N. A. A. Qasem, A. Peedikakkal, T. Laoui and M. Ali, Appl. Energy, 2016, 161, 225–255 CrossRef
- R. Khalilpour, K. Mumford, H. zhai, A. Abbas, G. Stevens and E. S. Rubin, J. Cleaner Prod., 2015, 103, 286–300 CrossRef CAS
- S. Y. Lee and S. J. Park, J. Ind. Eng. Chem., 2015, 23, 1–11 CrossRef CAS
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed
- The global status of CCS – 2015, Global Carbon Capture and Storage Institute, Melbourne, 2015 Search PubMed.
- F. Karadas, M. Atilhan and S. Aparicio, Energy Fuels, 2010, 24, 5817–5828 CrossRef CAS
- M. Ramdim, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 CAS
- Z. Lei, C. Dai and B. Chen, Chem. Rev., 2014, 114, 1289–1326 CrossRef CAS PubMed
- L. C. Tomé and I. M. Marrucho, Chem. Soc. Rev., 2016, 45, 2785–2824 RSC
- M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Sun, J. S. Thrasher, K. Kirichenko, S. Sinngh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554–2556 RSC
- T. P. T. Pham, C. W. Cho and Y. S. Yun, Water Res., 2010, 44, 352–372 CrossRef CAS PubMed
- B. Peric, J. Sierra, E. Marti, R. Cruañas, M. A. Garau, J. Aming, U. Bottin-Weber and S. Stolbe, J. Hazard. Mater., 2013, 261, 99–105 CrossRef CAS PubMed
- R. Biczak, B. Pawlowska, P. Balczewski and P. Rychter, J. Hazard. Mater., 2014, 274, 181–190 CrossRef CAS PubMed
- K. Paduszynski and U. Domanska, J. Chem. Inf. Model., 2014, 54, 1311–1324 CrossRef CAS PubMed
- L. Chen, M. Sharifzadeh, N. MacDowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC
- G. Cevasco and C. Chiappe, Green Chem., 2014, 16, 2375–2385 RSC
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. Silva-Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS
- G. García, S. Aparicio, R. Ullah and M. Atilhan, Energy Fuels, 2015, 29, 2616–2644 CrossRef
- T. J. Trivedi, J. H. Lee, H. J. Lee, Y. K. Jeong and J. W. Choi, Green Chem., 2016, 18, 2834–2842 RSC
- E. Ali, M. K. Hadj-Kali, S. Mulyono and I. Alnashef, Int. J. Greenhouse Gas Control, 2016, 47, 342–350 CrossRef CAS
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed
- Q. Zhang, K. De Oliveira-Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed
- B. Singh, H. Lobo and G. Shankarling, Catal. Lett., 2011, 141, 178–182 CrossRef CAS
- K. Radosevic, M. C. Bubalo, V. G. Srcek, D. Grgas, T. L. Dragicevic and I. R. Redovnikovic, Ecotoxicol. Environ. Saf., 2015, 112, 46–53 CrossRef CAS PubMed
- Q. Wen, J. X. Chen, Y. L. Tang, J. Wang and Z. Yang, Chemosphere, 2015, 132, 63–69 CrossRef CAS PubMed
- Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed
- G. García, M. Atilhan and S. Aparicio, Chem. Phys. Lett., 2015, 634, 151–155 CrossRef
- G. García, M. Atilhan and S. Aparicio, Int. J. Greenhouse Gas Control, 2015, 39, 62–73 CrossRef
- R. Ullah, M. Atilhan, B. Anaya, M. Khraisheh, G. García, A. Elkhattat, M. Tariq and S. Aparicio, Phys. Chem. Chem. Phys., 2015, 17, 20941–20960 RSC
- G. García, M. Atilhan and S. Aparicio, J. Mol. Liq., 2015, 211, 506–514 CrossRef
- G. García, M. Atilhan and S. Aparicio, J. Phys. Chem. C, 2015, 119, 21413–21425 Search PubMed
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2007, 9, 3397–3406 RSC
- X. Li and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 835–839 CrossRef CAS PubMed
- M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. O. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, A. D. N. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09. Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed
- E. W. Lemmon, M. L. Huber and M. O. McLinden, M.O. NIST Standard Reference Database 23: Reference Fluid Thermodynamic and Transport Properties-REFPROP, Version 9.1, National Institute of Standards and Technology, Standard Reference Data Program, Gaithersburg, 2013 Search PubMed
- A. P. Lyubartsev and A. Laaksonen, Comput. Phys. Commun., 2000, 128, 565–589 CrossRef CAS
- U. L. Essmann, M. L. Perera, T. Berkowitz, H. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS
- M. Tuckerman, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990–2001 CrossRef CAS
- V. Zoete, M. A. Cuendet, A. Grosdidier and O. Michielin, J. Comput. Chem., 2011, 32, 2359–2368 CrossRef CAS PubMed
- G. García, M. Atilhan and S. Aparicio, J. Phys. Chem. C, 2015, 119, 21413–21425 Search PubMed
- H. M. Lee, I. S. Youn, M. Saleh, J. W. Lee and K. S. Kim, Phys. Chem. Chem. Phys., 2015, 17, 10925–10933 RSC
- P. M. Gallagher, M. P. Coffey, V. J. Krukonis and N. Klasutis, in Supercritical Fluid Science and Technology, ed. K. P. Johnson and J. M. L. Penninger, American Chemical Society (ACS), Washington, D.C., 1989, ACS Symposium Series, vol. 406, chap. 22, pp. 334–354 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 (DFT results); Table S1 (systems used for MD simulations with CO2); Table S2 (forcefield parameterization using along this work); Fig. S2 (comparison between experimental and molecular dynamics predicted thermophysical properties); Fig. S3 (calculated residence times for CHCl_PhOAc_1_2); Fig. S4 (intermolecular interaction energies in CHCl_PhOAc_1_2); Fig. S5 (radial distribution functions in CHCl_PhOAc_1_2 + CO2); Fig. S6 (residence times in CHCl_PhOAc_1_2 + CO2); Fig. S7 (density profiles in CHCl_PhOAc_1_2 + CO2); Fig. S8 (snapshot for CHCl_PhOAc_1_2 + CO2); Fig. S9 (intermolecular interaction energy in CHCl_PhOAc_1_2 + flue gas). See DOI: 10.1039/c6ra22312e |
| This journal is © The Royal Society of Chemistry 2016 |