
The exclusive response of LSPR in uncapped gold nanoparticles towards silver ions and gold chloride ions†
Abstract
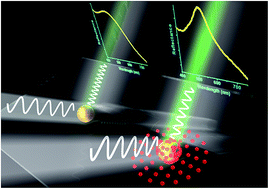
We report a significant modulation of the plasmon peak of a bare gold nanoparticle system after treatment with Ag+ or [AuCl4]− ions. The shift is highly selective to the presence of Ag+/[AuCl4]− metal ions and is not observed when other ions (Cu2+, Cd2+, Ce2+, Hg2+, Na+ and K+) are tested. The value of the red shift depends on the concentration of ions. As compared to Ag+ ions, the shift is larger in the presence of [AuCl4]− ions. We attribute the red-shift in plasmon to the formation of atomic clusters of Ag or Au and a change in relaxation times of hot electrons at the gold nanoparticle surface after treatment with Ag+ or [AuCl4]− ions. The red-shift is also sensitive to capping on gold nanoparticles, and silver ions do not shift the LSPR when gold nanoparticles are capped with citrate. The study of the effect of ions on the plasmon of AuNPs is significant from the viewpoint of designing highly selective AuNP based gold and silver ion sensors. The role of charge transfer and changes in electron dynamics, which lead to shifting of the localized surface plasmon resonance in AuNPs, has been exemplified as well. The versatility of the sensing approach has been tested in both solution-based and substrate-based configurations.
 Please wait while we load your content...
Please wait while we load your content...

