
Unique method to improve the thermal properties of bisphenol A tetraacrylate by graphite oxide induced space confinement†
Titash Mondal*ab,
Varunesh Chandrab and
Anil K. Bhowmick*a
aRubber Technology Center, Indian Institute of Technology Kharagpur, West Bengal, India 721302. E-mail: anilbhowmick@gmail.com; titash786@gmail.com
bDepartment of Chemistry, Indian Institute of Technology Patna, Bihar, India 800013
First published on 25th October 2016
Abstract
Enhancing the thermal stability of different epoxy acrylates is a critical step towards development of thermally stable organic coating materials. Herein, we examine the thermal properties of a model system based on bisphenol A tetraacrylate by a graphite oxide induced space confinement. Studies related to understanding the critical role of anisotropic particles like graphite and its derivatives in improving the thermal properties of commercially important different epoxy acrylates is still obscure. The samples with 2% (AP2) and 4% (AP4) of graphite oxide loading were found to significantly delay the onset of the thermal degradation of bisphenol A tetraacrylate by 17 °C and 28 °C respectively. Scanning electron microscopy and X-ray diffraction studies of the composites indicated that the bisphenol A tetraacrylate was intercalated between the graphite oxide sheets, resulting in the formation of a nano brickwall type super structure. The d-spacing of graphite oxide was noted to be 0.8 nm, while in the composite, the d-spacing value obtained for AP4 was 1.2 nm. Thermodynamic calculations indicated significant perturbation in the radius of gyration of the pre-polymer in the presence of graphite oxide sheet. Such a finding can be readily extended to other layered like filler materials to understand the physical properties of the pre-polymers under space confinement.
1. Introduction
The conjunction of graphene with different polymers for the development of advanced functional materials has attracted significant attention in recent times.1–3 The unique combination of the atomically thin dimensions, high strength and the ability to transport electrons ballistically makes graphene a wonder material.4–6 The incorporation of anisotropic graphene nanoplatelets is noted to affect the physico-chemical behavior of neat polymers. Modulation of the properties of neat polymers by the addition of graphene nanoplatelets is attributed to the positive interaction taking place between the nanoplatelets and the polymer chains. The surface chemistry of the nanoplatelets is crucial in determining the extent of interaction between the polymer chains and the filler materials. Incorporation of unfunctionalized graphene nanoplatelets often leads to the development of subtle interfaces and thereby imparting an inferior property to the nanocomposite.Accordingly, a priori designing of the functional groups on the surface of the graphene nanoplatelets is desirable for the development of a stable interface between the filler and polymer chains. For instance, addition of 0.1% modified graphene platelets is reported to enhance the tensile strength of the pristine polymer by ∼31%.7 Similarly, a potential improvement of the gas barrier property as well as the conductivity of the polyurethane was reported to take place upon addition of 1% of modified graphene nanoplatelets.8 Neat polymers are reported to register an inferior thermal stability. There is a strong correlation between the thermal stability and the service life of polymer nanocomposite. Thus, research related to the improvement of the thermal stability of the nanocomposite is demanding. The first such report on enhanced thermal stability of the polymer nanocomposite was demonstrated by Blumstein, wherein the thermal stability of the polymethyl methacrylate was found to be higher than the neat polymer due to the addition of layered silicate material.9 There has been a plethora of utilization of a different anisotropic and isotropic nanoparticles for enhancing the thermal stability of polymer matrix.10,11 Although a large volume of research has been dedicated to studying the thermal stability of layered-silicate/polymer composite, a few attempts have been made to test the potential of graphite oxide as a thermal stabilizer for the polymeric materials and also to understand the underlying mechanism involved in enhancing the thermal stability of the composite.
Even though there are multiple applications of polymer nancomposite, research related to the utilization of UV-curable nanocomposite adhesives and coating are gaining antecedence in the last few years.12–14 The low volatile organic content involved with these adhesives and coatings increases their importance. However, shortcomings like poor thermal stability of these epoxy acrylates often limit its usage for high temperature applications.15 A few attempts have been made to increase the thermal stability of the epoxy acrylate by combining them with silicone,16 blending the coating material with nitrogen and phosphorous containing compound,17 developing nanocomposites with zinc and aluminium nanoparticles;18 however, research related to study the effect of graphite oxide as a thermal stabilizer for epoxy acrylate coating material is far and few.19 It is worthy of mentioning that these acrylate pre-polymers are commonly considered as the building block for long chain polymers.
Thus, in light of this, we present a strategy, where graphite oxide was added to the pre-polymer. This was done to increase the thermal stability of the pre-polymer. It is conjectured that these graphite oxide–acrylate pre-polymer mixture can be further used to build thermally stable polymer. This makes the technique unique of its kind. Thus the novelty of the work is to prepare and characterize thermally stable graphite oxide based pre-polymer nanocomposites. Large number of oxygen functionalities on the surface of the graphite oxide promoted non-covalent interactions between the bisphenol A tetraacrylate and the graphite oxide. Such interactions resulted in improvement of the properties of nanocomposite over the neat bisphenol A tetraacrylate. To the best of our knowledge, study on such system is still obscure in the literature. The developed nanocomposites were characterized by different techniques.
2. Experimental section
2.1 Materials
Bisphenol A diglycidyl ether and acrylic acid were procured from Sigma Aldrich. Expanded graphite (grade 3777) was received as a gift from Asbury Carbon, NJ, USA. All other chemicals were procured from CDH Chemicals, India and used without further purification.2.2 Synthesis of acrylate pre-polymer
The synthesis of the prepolymer involved two steps. The first step involves the breaking of epoxy linkage present in the BADGE (bisphenol-A diglycidyl ether) with four equivalents of a sodium hydroxide in a methanolic solution. Briefly, 10 g BADGE (inhibitor free) was dissolved in a minimum amount of methanol. Into it, a solution of 4.7 g of sodium hydroxide dissolved in a minimum amount of methanol was added slowly under stirring condition at 30 °C and was allowed to stir for 1 h. In the second step, the sodium salt of bisphenol A-diglycidyl ether so formed was made to react with four equivalents of acrylic acid (8.1 ml). The mixture was further left under stirring for 1 h. A white-sticky precipitate was obtained, which was re-precipitated in dichloromethane. The supernatant containing low molecular weight fractions and un-reacted molecular species were discarded and the remaining suspension was centrifuged at 2000 rpm for four minutes and the residual compound were vacuum dried until the compound got rid of the solvent. The yield of the reaction was 82%.2.3 Synthesis of graphite oxide
2 g expanded graphite was taken, to which 46 ml of concentrated H2SO4 was added slowly with constant stirring at a temperature less than 5 °C. This was followed by the addition of 6 g of KMnO4 very slowly, in small amounts, with constant stirring and it was stirred for two hours afterwards at a temperature less than 5 °C. A slurry so obtained was stirred for an hour at 35 °C. The slurry was diluted further with 92 ml of deionized water and the temperature of the reaction was raised to 90 °C. 280 ml of deionised water and 12 ml of 50% H2O2 was added to the mixture, resulting in a color change of the suspension from dark brown to yellowish brown. The slurry product obtained was washed with 10% HCl to make the product free of sulfate ions and was further washed with deionized water until the pH became neutral. The centrifugation was performed at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The product was vacuum dried for further characterization and use.
000 rpm. The product was vacuum dried for further characterization and use.
2.4 Preparation of the bisphenol A tetraacrylate–graphite oxide composite
Bisphenol A tetraacrylate–graphite oxide composite with 2%, 4% and 6% filler loading were prepared. 0.5 g of BADGE acrylate pre-polymer was dispersed in a minimum amount of water while, in a separate container, different amounts of graphite oxide (10 mg GO for 2% loaded sample, 20 mg GO for 4% loaded sample, and 30 mg GO for 6% loaded sample) were ultrasonicated for 1 h in water. The dispersed graphite oxide was further added to the acrylate pre-polymer solution in water, and was allowed to stir for three hours at room temperature. The suspension was transferred to a glass Petri dish and was left undisturbed for ten hours after which it was vacuum dried. The isolated composite materials were further characterized.2.5 Characterization
The Fourier transform infrared (FTIR) spectroscopy of the synthesized bisphenol A tetraacrylate, graphite oxide and the nanocomposites were recorded on a PerkinElmer 400 FTIR instrument, adopting attenuated total reflectance mode with a resolution of 2 cm−1. The thermal stability of the bisphenol A tetraacrylate and the nanocomposite was recorded by thermogravimetric analysis (TGA) in a nitrogen atmosphere from room temperature to 800 °C. A ramp rate of 10 °C min−1 was maintained throughout the experiment (SD Q600 TA Instruments, U.S.A.). Raman spectroscopy for the bisphenol A tetraacrylate was examined using the STR 750 series Raman spectrometer (Technos Instrument, India) A 514.5 nm Ar-ion-laser source and grating of 600 lines per mm was used for the experiment. The morphology of the graphite oxide was monitored using transmission electron microscope (TEM) (JEM 2100 JEOL) and that for nanocomposite was observed using a field emission scanning electron microscopy (FESEM, ZEISS MERLIN GEMINI 2).3. Results and discussion
3.1 Synthesis and characterization of bisphenol A tetraacrylate
Bisphenol A tetraacrylate was synthesized as shown in Scheme 1. At the first step of the reaction, methanolic solution of sodium hydroxide was added slowly. On the successful opening of the epoxy ring, acrylic acid was added to the sodium salt of bisphenol A diglycidyl ether. The reaction was performed in two steps to avoid a side reaction between sodium hydroxide and acrylic acid. In order to support the fidelity of the scheme, FTIR spectra of the reaction mixture at different time intervals of the reaction were studied. It was observed that the 910 cm−1 peak, which corresponds to the epoxy linkage decreased with the reaction time. A complete cleavage of the epoxy linkages was reflected from the disappearance of the epoxy peak with the progress of time (Fig. S1†).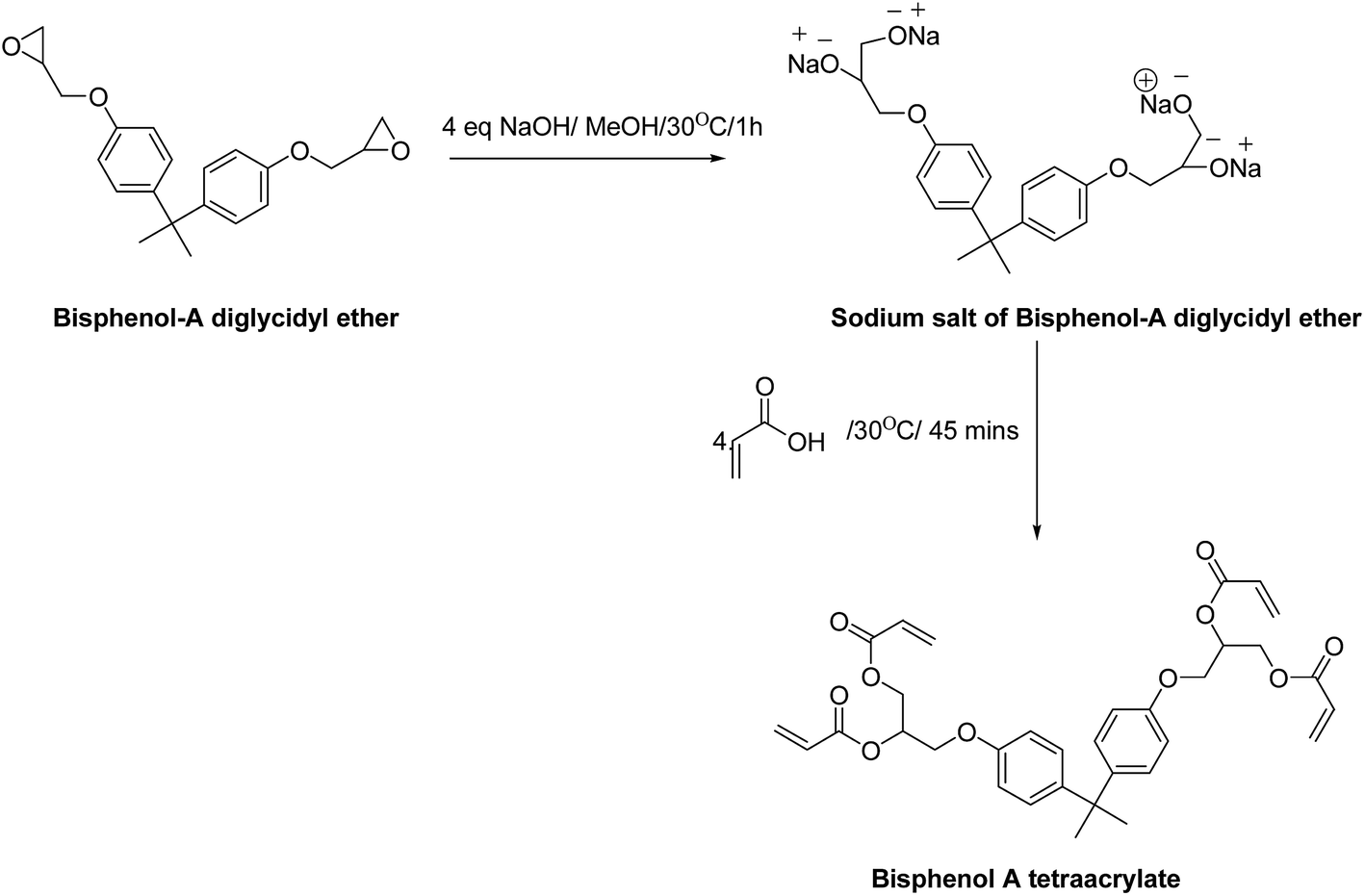 | ||
| Scheme 1 Synthesis of bisphenol A tetraacrylate from bisphenol-A diglycidyl ether and acrylic acid. | ||
The addition of four parts of acrylic acid to this sodium salt resulted in the formation of the bisphenol A tetraacrylate or the acrylate based pre-polymer (AP). A sharp peak was noted at 1723 cm−1 due to the carbonyl group of the pre-polymer (Fig. S2†). A peak at 1635 cm−1 in the pre-polymer spectrum affirmed the presence of acrylate, which was absent in the spectrum of BADGE, as shown in Fig. S2.† The sharp peaks at 660 cm−1, 836 cm−1, 898 cm−1, 950 cm−1 and 988 cm−1 corresponded to the C–H out of plane bending in the pre-polymer spectrum and the peaks at 1437 and 1510 cm−1 due to CH3– bend and the –CH2– bending respectively were observed. The peak at 1546 cm−1 is a resultant of the ‘in-ring’ C–C stretching in the aromatic group; the peaks at 1280 cm−1, 1248 cm−1, 1183 cm−1, 1132 cm−1, 1106 cm−1, 1053 cm−1 and 1039 cm−1 confirmed the presence of ester and ethereal linkage in the pre-polymer. Additionally, the Raman spectrum of the pre-polymer (Fig. S3†) showed strong Raman shifts at 1639 cm−1, 1461 cm−1 and 1288 cm−1 corresponding to ν(C–C) aromatic ring chain vibrations, δ(CH2) and δ(CH3) asymmetry, and ν(C–C) aliphatic chain vibrations respectively.
3.2 Synthesis and characterization of graphite oxide
Graphite oxide (GO) was synthesized using the technique due to Hummers.20 The FTIR measurement of the graphite oxide showed the peaks at 1016 cm−1, 1032 cm−1 and 1055 cm−1 that affirmed the presence of the carboxylic acid groups in the compound (Fig. S4†). The 1225 cm−1 peak indicated to the C–O stretching of alcohols and carboxylic acids; the peak at 3193 cm−1 showed the presence of hydroxyl groups. C–H rocking of the alkyl was observed at 1371 cm−1. The peak at 1618 cm−1 corresponded to ‘in ring’ aromatic C–C stretching. The peak at 1720 cm−1 reflected the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carboxylic acids. The peaks from 2844 cm−1 to 2985 cm−1 were due to C–H stretching of the alkyl chains. The peaks in the range of 3600 cm−1 confirmed the presence of free –OH groups. Additionally, the graphite oxide demonstrated a good electron transparency compared to the starting material, expanded graphite (as shown in Fig. 1a and b). Micro corrugation and ripples over the surface of GO were observed. Wide angle X-ray diffraction of GO (Fig. S5†) indicated a d-spacing of 0.8 nm of the [001] plane. The observation in TEM and d-spacing value obtained is in line with the existing literature.21,22 The size of the expanded graphite and graphene oxide, as corroborated from the dynamic light scattering experiment was noted to be 275 nm and 126 nm respectively.
O stretching of the carboxylic acids. The peaks from 2844 cm−1 to 2985 cm−1 were due to C–H stretching of the alkyl chains. The peaks in the range of 3600 cm−1 confirmed the presence of free –OH groups. Additionally, the graphite oxide demonstrated a good electron transparency compared to the starting material, expanded graphite (as shown in Fig. 1a and b). Micro corrugation and ripples over the surface of GO were observed. Wide angle X-ray diffraction of GO (Fig. S5†) indicated a d-spacing of 0.8 nm of the [001] plane. The observation in TEM and d-spacing value obtained is in line with the existing literature.21,22 The size of the expanded graphite and graphene oxide, as corroborated from the dynamic light scattering experiment was noted to be 275 nm and 126 nm respectively.
 | ||
| Fig. 1 Selective area TEM micrograph of (a) expanded graphite and (b) graphite oxide. | ||
3.3 Preparation of the bisphenol A tetraacrylate–graphite oxide composite and its characterization
A solution mixing methodology was adopted for the development of graphite oxide–acrylate pre-polymer composite at different filler concentrations (2, 4 and 6%; AP2, AP4 and AP6). The developed composite demonstrated conglomeration of typical peaks corresponding to the neat pre-polymer and the graphite oxide (as discussed in the Sections 3.1–3.2). No new additional peaks were noted for the developed composites. A detailed discussion related to the shift in the peak position noted in the FTIR spectra for the developed composite is discussed in the later section of the manuscript.The thermal stability of the neat pre-polymer and the nanocomposite so prepared was determined using thermogravimetric analysis (TGA) in a nitrogen atmosphere (Fig. 2). The neat pre-polymer registered a two step degradation. The first onset of degradation was noted at 266 °C, whereas the second onset of degradation was noted at 418 °C. The first step of degradation was attributed to the degradation of the bisphenol-A diglycidyl ether unit and the second step was due to the acrylic unit. Interesting changes in the thermal degradation profile were noted upon addition of graphite oxide. The first step of degradation of the polymer nanocomposite was marginally affected in the presence of graphite oxide. However, a significant delay in the degradation of the acrylic part was noted with the addition of graphite oxide. The addition of 2% and 4% of graphite oxide shifted the degradation onset temperature by 17 °C and 28 °C respectively, while the temperature at which maximum degradation took place for AP2 and AP4 were found to be shifted by 6 °C and 10 °C respectively (as shown in the first derivative plot in the inset of Fig. 2). However, the AP6 registered an onset of degradation of the acrylic unit around 420 °C. Additionally from the residual percentage obtained from the TGA experiment, it can be reasonably inferred that the flame resistivity of the nanocomposite was significantly increased compared the neat acrylate pre-polymer. Using the equation due to van Krevelen,23 the limited oxygen index (LOI) value for the neat acrylate pre-polymer, AP2, AP4 and AP6 was estimated. The fire retardant nature of any polymeric material can be correlated to the change in critical concentration of oxygen that they induce as a function of their concentration and is commonly expressed as limiting oxygen index (LOI):
| LOI = [O2]/[O2] + [N2] | (1) |
 | ||
| Fig. 2 TGA traces of AP, AP2, AP4 and AP6 respectively, recorded in a nitrogen atmosphere from room temperature to 800 °C at a ramp rate of 10 °C min−1. Inset shows the first derivative plot of the degradation pattern. | ||
Even though, AP2 and AP4 demonstrated an increasing trend in the thermal stability, AP6 registered inferior stability compared to the other two nanocomposite. Such a behavior can be correlated to the dispersion of nanofiller and the van der Waals interaction among the filler materials inside the acrylate pre-polymer matrix. The van der Waals interaction (W) acting between parallel plate filler material25,26 can be expressed as
 | (2) |
It is worthy to note that the Gaussian simulation is an effective pathway to probe the polymer–filler interaction. Perez et al. demonstrated the interaction of elastomers (by using small molecules) with silica nanoparticles using Gaussian simulation.27 On a similar note, the geometry optimization of the bisphenol A tetraacrylate was done using the Gaussian package with the Becke's three-parameter hybrid functional (B3LYP) method. 6-311G (d, p) was used as the basis set. As shown in Fig. 3, bisphenol A tetraacrylate demonstrated a twisted morphology with protruding carbonyl atoms of the acrylate moiety. Thus, during the intercalation process, the carboxylic group and the hydroxyl group of GO will interact strongly via non-covalent interactions with the acrylate unit. This will result in the acrylate section of the pre-polymer to behave as a stiff unit and hence the onset of the degradation of the acrylate unit will be significantly affected in the presence of GO. The bisphenol part of the pre-polymer exhibits a twisted structure around the C–C bond connecting the two benzene rings. As a result, interaction of graphite oxide through π–π stacking with a benzene ring of bisphenol unit is largely affected. Thus, the onset of degradation of the bisphenol unit was marginally affected.
 | ||
| Fig. 3 Gaussian optimized ground state structure of acrylate pre-polymer. The white balls corresponds to the hydrogen atom, red balls corresponds to the oxygen atom while the gray balls correspond to the carbon atom. | ||
In an attempt to support the above hypothesis about the dispersion of the nanofiller inside the pre-polymer matrix, the structure of the composites was analyzed using high resolution X-ray diffraction technique (XRD) and was compared with that of the neat pre-polymer. The neat pre-polymer demonstrated a crystalline peak at 2θ = 8.5° (Fig. 4). However, peak maxima for the pre-polymer appeared to be shifted to higher 2θ in the presence of graphite oxide, thereby indicating about significant structural perturbation of the crystal packing of the pre-polymer. In the case of AP2, the signature peak for GO was not decipherable due to filler loading. Nevertheless, the peak for GO was noted in the case of AP4 and AP6. The striking difference in the peak position of GO was noted compared to the neat GO. The peak maxima for GO in the case of AP4 was observed at 2θ = 7.1, whereas the 2θ shifted to 7.9° for AP6 (Fig. 4). The d-spacing value obtained for AP4 and AP6 were 1.2 nm and 1.1 nm respectively. This indicated that the GO platelets were more intercalated in the case of AP4 compared to AP6. As a result, a higher degree of space confinement of the pre-polymer chains for AP4 over AP6 can be anticipated. Such an observation about better degree of dispersion of the GO inside the pre-polymer matrix can be correlated with the dispersion of the nanofillers in the polymer matrix and is in line with the existing literature.
 | ||
| Fig. 4 Wide angle X-ray diffraction of pre-polymer (AP), AP2, AP4 and AP6 respectively, using Cu Kα and λ = 0.154 nm. | ||
Additionally, on careful evaluation of the TEM micrograph of the GO, it can be seen than micro wrinkles are present on the surface. This surface of GO with micro-corrugated surface acts as binding site for the attachment of the pre-polymer. As a result, better interaction between the pre-polymer and the filler is achieved. Through a combination of intercalation of the bisphenol A tetraacrylate inside the gallery spacing of GO (from XRD) and through physical anchorage of the bisphenol A tetraacrylate, it is conjectured that a nanobrick wall super structure was generated.
Further, attempts were made to understand the nature of the filler arrangement in the pre-polymer composite. Owing to the best properties noted for AP4, it was selected for microscopic analysis using field emission scanning electron microscopy. As shown in Fig. 5a and b, the GO platelets were well dispersed in the pre-polymer (surface view). On a careful evaluation of the vertical cross-section of the sample, it was observed that the GO platelets were hierarchically arranged inside the matrix in a layered pattern (Fig. 5c and d).
 | ||
| Fig. 5 FE-SEM micrograph (a and b) of AP4 recorded on the surface at 10k and 20k magnification (scale bar 2 μm) and of (c and d) vertical cross-section of AP4 demonstrating layered like arrangement recorded at 20k and 35k magnification (scale bar 1 μm). | ||
An insight towards the interaction between the filler material and the polymer chains can be substantiated using FTIR spectroscopy.26 The polymer composite at different filler loadings were analyzed in an ATR mode (3 mg of sample was taken in each case). The carbonyl peak of the pre-polymer was selected as the benchmark to estimate the interaction between the filler material and the polymer chain. As shown in Fig. 6a, the peak due to carbonyl stretching is progressively broadening for AP2 and AP4 compared to the neat pre-polymer. However, the peak of the carbonyl group for AP6 was marginally broadened than that of the neat pre-polymer. This indicated that at 2% and 4% filler loading, the functional group of the GO and the carbonyl group of the acrylic moiety interacted through hydrogen bonding. As a result, broadening of the peak of the carbonyl group was noted. Such an observation is in line with the prediction made from TGA experiment.
 | ||
| Fig. 6 Selective area FTIR spectra for AP, AP2, AP4 and AP6 respectively, showing the (a) peak broadening of the carbonyl peak and (b) shift in the peak due to ester linkages. 64 scans were taken in the ATR mode. | ||
A shift in the peak position of the FTIR can be correlated with the pre-polymer–filler interaction as well as the thermodynamic feasibility of the nanocomposite formation.28 The peak due to the ethereal linkage of the pre-polymer at 1236 cm−1 was found to be shifted to higher wavenumber (Fig. 6b). The peaks were noted at 1252 cm−1, 1249 cm−1, 1247 cm−1 respectively for AP4, AP2 and AP6. This indicates about substantial interaction between the oxygen functionalities of the filler materials with that of the bisphenol A tetraacrylate. The interaction between bisphenol A tetraacrylate and nanoparticle can be explained in terms of a mean-field thermodynamic model.
The free energy change in polymer under graphene confinement can be expressed as
| ΔGAP = ΔHAP − TΔSAP | (3) |
| ΔGGO = ΔHGO − TΔSGO | (4) |
| ΔGCom = ΔHCom − TΔSCom = ΔHCom − T(ΔSAP + ΔSGO) | (5) |
For a thermodynamically favorable process, the value of ΔGCom should be negative. Intercalation of the bisphenol A tetraacrylate in between graphite oxide platelets decreases the ΔSAP as the degrees of randomness decreases. However, ΔSGO value increases due to the exfoliation of the graphene sheet. Thus, ΔHCom value becomes the controlling parameter for favorable reaction. The shift in the peak position can be correlated with the ΔHCom value using the Fowke's equation29,30 as given by
| ΔH = 0.236 × Δν | (6) |
The Δν value corresponds to the shift in the peak position of the ethereal linkages for the nanocomposite compared to the pure polymer. The ΔH values for AP6, AP4 and AP2 were −2.6 kcal mol−1, −3.8 kcal mol−1 and −3 kcal mol−1 respectively. Thus, from this thermodynamic data obtained, it can be reasonably inferred that the formation of AP4 was most favourable compared to AP2 and AP6. Such an observation is in line with the observation made from XRD as well as the predictions made from thermal analysis.
The effect on the radius of gyration (Rg) of the polymer due to the addition of nanoparticle into the polymer matrix is a matter of much debate.31 Both increase as well as decrease in the Rg have been reported to take place in the presence of the nanoparticles.32 However, in the present case, a relative estimation about the Rg of the pre-polymer in the presence of graphite oxide was done by utilizing the empirical formula proposed by Sanchez33
 | (7) |
Incidentally, the perturbation of the polymer induced by the GO sheet is also bound to affect the transition dynamics of the confined polymer chains. Zhang et al. reported that (i) size effect and (ii) interfacial effect is the contributory factors controlling the transitions under confinement.34 Representative sample, AP4 was subjected to a dynamic mechanical condition. In the window of the study, one of such prominent transition of AP4 (−44 °C) was lower than that of AP (−35 °C) (as shown in Fig. S6†).
4. Concluding remarks
In conclusion, we have presented a strategy, where graphite oxide was added to the acrylate pre-polymer to increase the thermal stability of the latter. It is conjectured that these graphite oxide–acrylate pre polymer mixture can be further used to build different kinds of thermally stable polymers. This makes the technique unique of its kind. Further, we have provided an insight about the critical role of graphite oxide to act as a thermal stabilizer for the bisphenol A tetraacrylate (acrylate based pre-polymers). Structural elucidation through theoretical studies indicated that such a structure of the pre-polymer unequivocally assisted in the formation interlocking with micro-corrugation/ripples on the surface of graphite oxide. The graphite oxide platelets acted as a thermal shield for the pre-polymer and thereby increased the thermal stability of AP2 and AP4 respectively. Such an increase in the thermal stability was found to be related to the concentration of the nanophase. It was noted that with an increase in the filler concentration to 6% facilitated the filler–filler, van der Waals and other non-covalent interactions. This causes the formation of large sized agglomeration. As a result, it could be conjectured that the filler–polymer interaction could not compensate the filler–filler interaction and hence an inferior thermal stability for AP6 was registered over AP2 and AP4. Further, the Rg value was found to decrease for AP4 and AP2 under confinement. It would be interesting in the future to study the chain relaxation process for such acrylate pre-polymers under graphene induced space confinement.Acknowledgements
T. M. and A. K. B. appreciate the partial support of Birdgestone Corporation, Japan.References
- T. Mondal, R. Ashkar, P. Butler, A. K. Bhowmick and R. Krishnamoorti, ACS Macro Lett., 2016, 5, 278–282 CrossRef CAS.
- R. M. Santos, C. Vilaverde, E. Cunha, M. C. Pavia and J. A. Covas, Soft Matter, 2016, 12, 77–86 RSC.
- M. Tokuda, M. Yamane, S. C. Thickett, H. Minami and P. B. Zetterlund, Soft Matter, 2016, 12, 3955–3962 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- T. Mondal, A. K. Bhowmick and R. Krishnamoorti, Chem. Mater., 2015, 27, 713–725 CrossRef.
- T. Mondal, A. K. Bhowmick and R. Krishnamoorti, ACS Appl. Mater. Interfaces, 2014, 6, 7244–7253 CAS.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef CAS PubMed.
- H. Kim, Y. Miura and C. W. Macosko, Chem. Mater., 2010, 22, 3441–3450 CrossRef CAS.
- A. Blumstein, J. Polym. Sci., Part A: Polym. Chem., 1965, 3, 2665–2672 CAS.
- A. Choudhury, A. K. Bhowmick and C. Ong, J. Appl. Polym. Sci., 2010, 116, 1428–1441 CAS.
- S. D. Burnside and E. P. Giannelis, Chem. Mater., 1995, 7, 1597–1600 CrossRef CAS.
- M. A. Osman, V. Mittal, M. Morbidelli and U. W. Suter, Macromolecules, 2003, 36, 9851–9858 CrossRef CAS.
- W. O. Gordon, G. W. Peterson and E. M. Durke, ACS Appl. Mater. Interfaces, 2015, 7, 6402–6405 CAS.
- S. Qiu, S. Li, Y. Tao, X. Feng, B. Yu, X. Mu, W. Xing, Y. Hu and G. Jie, RSC Adv., 2015, 5, 73775–73782 RSC.
- Y.-J. Park, D. H. Lim, H.-J. Kim, D.-S. Park and I.-K. Sung, Int. J. Adhes. Adhes., 2009, 29, 710–717 CrossRef CAS.
- M. Bajpai, V. Shukla and F. Habib, Prog. Org. Coat., 2005, 53, 239–245 CrossRef CAS.
- Y. Tan, Z.-B. Shao, X.-F. Chen, J.-W. Long, L. Chen and Y.-Z. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 17919–17928 CAS.
- C. Sow, B. Riedl and P. Blanchet, J. Coat. Technol. Res., 2011, 8, 211–221 CrossRef CAS.
- B. Yu, X. Wang, H. Yang, L. Song and Y. Hu, Ind. Eng. Chem. Res., 2012, 51, 14629–14636 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- D. W. van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- T. Kashiwagi, F. Du, K. I. Winey, K. M. Groth, J. R. Shields, S. P. Bellayer, H. Kim and J. F. Douglas, Polymer, 2005, 46, 471–481 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, San Diego, CA, 3rd edn, 2011 Search PubMed.
- T. Mondal, A. K. Bhowmick and R. Krishnamoorti, ACS Appl. Mater. Interfaces, 2014, 6, 16097–16105 CAS.
- L. D. Perez, E. Florez, J. E. Mark and B. L. Lopez, Polym. Int., 2009, 58, 811–816 CrossRef CAS.
- C. Mitchell and R. Krishnamoorti, Macromolecules, 2007, 40, 1538–1545 CrossRef CAS.
- F. M. Fowkes, D. O. Tischler, J. A. Wolfe, L. A. Lannigan, C. M. A. John and C. M. J. Halliwell, J. Polym. Sci., Part A: Polym. Chem., 1984, 22, 547–566 CrossRef CAS.
- N. Roy and A. K. Bhowmick, J. Phys. Chem. C, 2012, 116, 8763–8772 CAS.
- R. Krishnamoorti and R. A. Vaia, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 3252–3256 CrossRef CAS.
- A. Karatrantos, N. Clarke, R. J. Composto and K. I. Winey, Soft Matter, 2015, 11, 382–388 RSC.
- I. C. Sanchez, Macromolecules, 1979, 12, 980–988 CrossRef CAS.
- C. Zhang, Y. Guo and R. D. Priestley, Macromolecules, 2011, 44, 4001–4006 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra for pre-polymer and GO, Raman spectra for pre-polymer, TGA curve for the pre-polymer and BADGE is provided. Tan delta plot obtained from DMA. See DOI: 10.1039/c6ra22252h |
| This journal is © The Royal Society of Chemistry 2016 |