
A simple and efficient in situ generated ruthenium catalyst for chemoselective transfer hydrogenation of nitroarenes: kinetic and mechanistic studies and comparison with iridium systems†
Abstract
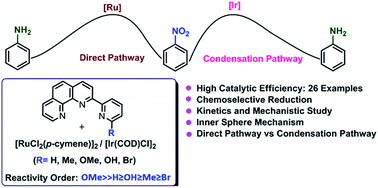
The catalytic activities of a series of in situ generated homogeneous ruthenium systems based on commercially available [RuCl2(p-cymene)]2 and various ligands in transfer hydrogenation of nitroarenes to anilines were investigated. Combination of [RuCl2(p-cymene)]2 and tridentate phenanthroline based ligand 2-(6-methoxypyridin-2-yl)-1,10-phenanthroline (phenpy-OMe) exhibited the highest catalytic activity for this reaction using 2-propanol as hydrogen source. This protocol provides a facile route to access aromatic amines under mild conditions in excellent yields. Notably, this system chemoselectively reduced the nitro groups over an array of other reactive functionalities such as ketone, alkene, amide, nitrile, and aryl halide. Operational simplicity, high yields, mild reaction conditions and short reaction times make this an attractive methodology for accessing various functionalized anilines. A series of controlled experiments and careful mechanistic investigation with the possible intermediates suggested that transformation of nitrobenzene to aniline with ruthenium and iridium system proceeded via direct route and condensation route respectively.
 Please wait while we load your content...
Please wait while we load your content...

