
Features of partial encapsulation of an ESIPT probe 3-hydroxy-2-naphthoic acid (3HNA) in the nano cavities of β- and γ-cyclodextrin: comparative study with sequestered 3HNA in micelles and reverse micelle†
Abstract
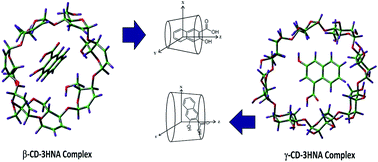
The role of intramolecular H-bonding within an excited state intramolecular proton transfer (ESIPT) probe, the intermolecular H-bonding ability of the microenvironment with the probe in different organized assemblies, the polarity of the medium after encapsulation and the motional restriction of the probe have been investigated by monitoring the ESIPT emission as well as the localized π–π* emission of 3-hydroxy-2-naphthoic acid (3HNA) in various confined media like cyclodextrins, micelles and reverse micelle. The steady state and time-resolved fluorescence and also the time resolved anisotropy measurements of the encapsulated probe indicate the formation of a 1 : 1 complex between the β- and γ-cyclodextrin hosts and the guest. The values of the binding constants and different thermodynamic parameters for complexation have also been reported. The results in various confined media have been correlated using control experiments on the ESIPT emission of 3HNA in mixed solvents consisting of dimethyl sulfoxide (DMSO) and water (mixture of an aprotic polar and a protic polar solvent) and also of acetonitrile (ACN) and dichloromethane (DCM) (mixture of an aprotic polar and a non-polar solvent). Theoretical calculation on the orientation of 3HNA within β- and γ-cyclodextrins has been carried out in order to explain the observed rotational correlation time of the probe in confined environments.
 Please wait while we load your content...
Please wait while we load your content...

