
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzeotrope enabled polymerization of ethylene oxide
Jennifer L.
Bento
a,
Drona R.
Madugula
b,
Chetan C.
Hire
a and
Douglas H.
Adamson
*ab
aInstitute of Materials Science, Polymer Program, University of Connecticut, Storrs, CT 06269, USA. E-mail: adamson@UConn.edu
bDepartment of Chemistry, University of Connecticut, Storrs, CT 06269, USA
First published on 4th October 2016
Abstract
Poly(ethylene oxide) (PEO) is a nonionic hydrophilic polymer having the same repeat unit as poly(ethylene glycol) (PEG), distinguished from PEG only by mass or synthetic approach. It is of interest in both biology and materials science, as PEO surfaces demonstrate a unique lack of protein adhesion and PEO block copolymers are widely used in applications such as drug delivery. However, the synthesis of PEO can be experimentally challenging, requiring air sensitive organometallic reagents to form reactive potassium alkoxides followed by the removal of compounds such as naphthalene from the final product. Here we report a synthetic route that avoids these difficulties by forming the propagating alkoxides by azeotropic distillation, removing water from the alcohol/alkoxide equilibrium. Removing the water drives the equilibrium to the potassium alkoxide without the use of pyrophoric organometallics. GPC and NMR are used to characterize the PEO polymers made by this approach from various alcohols, including hydroxyl terminated PEO.
Introduction
Poly(ethylene oxide) (PEO), also known as poly(ethylene glycol) (PEG), is a water soluble,1 nonionic semi-crystalline polymer2 that is biocompatible,3 nontoxic,4 and chemically stable.3 Due to PEO's ability to inhibit the adhesion of proteins, polymers containing PEO have a range of applications that include polymeric surfactants,5 emulsifiers,6 drug carriers,7–11 and anti-fouling coatings for medical implants12,13 and ship hulls.14 The laboratory synthesis of these materials, however, typically requires the use of moisture sensitive, pyrophoric organometallics in order to convert hydroxyl functional groups to the metal alkoxides required for the polymerization of ethylene oxide (EO). In this article we introduce a new mechanism for the formation of the necessary alkoxides from alcohols and hydroxyl functionalized polymers: the use of azeotropic distillation to remove water from the equilibrium between alcohol/KOH and alkoxide/water, as illustrated in Scheme 1. This approach uses KOH as the source for potassium cations, avoiding the use of moisture sensitive reagents that often need to be synthesized immediately prior to use.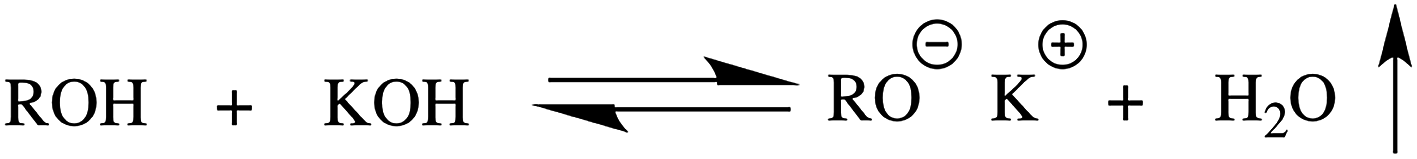 | ||
| Scheme 1 Equilibrium responsible for forming potassium alkoxide initiator. The removal of water drives the equilibrium to the right. | ||
Industrially, PEO homopolymers are synthesized at high temperatures and pressures (100–200 °C and 520 kPa) by adding ethylene oxide (EO) to an alcoholic aqueous solution containing a caustic.15 For lab scale synthesis, the anionic ring-opening polymerization (ROP) of EO is typically employed, with the initiator being a potassium alkoxide.3,16 Potassium, rather than lithium, is normally used due to the strong association between the lithium cation and the propagating oxygen anion17–19 that results in tight ion pairs and little to no chain propagation.17,18 This inability of lithium cations to propagate the growth of PEO means that adding EO to an anion with a lithium counter ion results in only one monomer unit being added to the chain. This is a useful approach to form hydroxyl terminated polymers, and is often used as a route to the polymers from which PEO is subsequently grown.
The synthesis of PEO polymers typically begins by reacting a hydroxyl group with an alkyl or aromatic potassium organometallic to form a potassium alkoxide. The compounds most frequently used are cumylpotassium,20,21 diphenylmethyl potassium (DPMK),22–24 benzyl potassium,1,25,26 α-phenyl ethyl potassium,27 and potassium naphthalenide.6,28 Examples include work by Allgaier et al., where cumylpotassium was added to hydroxyl terminated polymers to initiate the polymerization of EO to form poly[1,4-isoprene-b-(ethylene oxide)] (PI-PEO) and poly[ethylene-co-propylene-b-(ethylene oxide)] (PEP-PEO) block copolymers.20 As is common for such potassium reagents, the cumylpotassium had to be synthesized and used within a short period of time.20 Castle et al. synthesized PEO block copolymers using DPMK formed with potassium naphthalene.23 DPMK was also used to grow the PEO grafts in poly(styrene)comb-b-poly(ethylene oxide)comb copolymers,22 and as is often the case, PEO homopolymer was found as an impurity. Benzyl potassium has also been used for the polymerization of EO in the synthesis of PEO homopolymers, polystyrene-b-poly(ethylene oxide) (PS-b-PEO), and for PI-b-P2VP-b-PEO copolymers.1 α-Phenyl ethyl potassium has been used to synthesize PS-b-PEO and PS-b-PEO-b-PS copolymers27 with high conversion of EO (95%), but purification of the final product was necessary to remove PS and PEO homopolymers. Lastly, potassium naphthalide has been used in the synthesis of polyolefin-PEO block copolymers6 by forming the potassium alkoxide of hydrogenated polydienes containing hydroxyl end groups. Although this is far from a complete review of PEO block copolymer synthesis, these investigations are representative of the typical current approaches.
Other, much less common approaches have also been employed. Potassium methoxide was used to synthesize PS-b-PEO block copolymers,29 requiring a reaction time of 11 days, heating gradually from 30 °C to 70 °C for 7 days, followed by holding the reaction at 70 °C for another 4 days followed by the removal of PS and PEO homopolymers. Potassium metal has also been used directly to create potassium alkoxide initiators. In one example, potassium metal was added piecewise to a reaction mixture containing dimethylaminoethanol to form the potassium alkoxide.30 The addition of ethylene oxide was then followed by butylene oxide to produce a copolymer after 20 days. Another example of using potassium metal is the use of a potassium mirror to synthesize PS-b-PEO copolymers from hydroxyl terminated PS.31
Methods for growing PEO that do not involve potassium have been reported, but they are the exception. One route to PEO containing block copolymers allows for the use of lithium as a counterion by adding a phosphazene base.18,32 The phosphazene base complexes with the lithium counterion and allows for the propagation of EO. Polybutadiene-PEO (PBd-PEO) and PI-PEO block copolymers have been synthesized using this approach.18 Another route used a lithium alkoxide with small amounts of a potassium alkoxide in a benzene/DMSO mixture. PEO homopolymer was found in the final block copolymer product, but other alkoxides, such as potassium 2,6-di-t-butylphenoxide, produced less PEO homopolymer.33 To avoid the presence of potassium in the final material, an N-heterocyclic carbene was used to initiate EO polymerization, followed by the sequential polymerization of ε-caprolactone.3 Finally, cryptands have been used to complex the lithium ion and increase the reactivity of the anion. Block copolymers containing PBd and PI have been prepared by this approach.16,18,32,34
A common theme in nearly all of these methods is the conversion of an alcohol to a metal alkoxide by way of a reactive organometallic. In our approach, rather than adding a reactive and air sensitive pyrophoric compound to form the necessary potassium alkoxide, we add potassium hydroxide without any need for moisture-free conditions or prior synthesis of the organometallic. Additionally, at the end of the reaction, there are no compounds, such as naphthalene, that must be removed. The equilibrium between alcoholic potassium hydroxide and a potassium alkoxide, as shown in Scheme 1, generally favors the reactants. By Le Chatelier's principle, removal of the water from the right hand side drives the equilibrium towards the potassium alkoxide, eliminating the need for the addition of organometallic reagents or potassium metal. A typical reaction starts by dissolving the hydroxyl-containing molecule in toluene and adding a stoichiometric amount of KOH dissolved in methanol. The toluene is then partially distilled off, observing the boiling temperature of the toluene. Initially, the boiling temperature is far lower than the standard boiling temperature for toluene, as first the methanol, then the water in the form of an azeotrope with toluene, are removed. Once all the water in the system is removed, the boiling temperature reaches the literature value for toluene. After complete removal of the water, dry THF is added to provide a polar solvent for chain propagation, followed by the addition of the EO monomer. As examples of this approach we discuss: growing PEO off small molecular weight alcohols, extending the chain length of a PEO homopolymer, and synthesizing a block copolymer of polystyrene and PEO (PS-b-PEO) from a hydroxyl terminated PS.
Experimental
Materials
Cyclohexane, benzophenone, and tetrahydrofuran (THF) were purchased from Fisher. 0.1 N potassium hydroxide in methanol, 1.3 M sec-butyllithium in cyclohexane/hexane (92/8), 1,1-diphenylethylene (98%), styrene (99.5%), calcium hydride (93%), N,N-dimethylacetamide (99%), and chloroform-d (99.8 atom% D, 1 v/v% TMS) were purchased from Acros Organics. Methanol (≥99.8%), toluene (≥99.5%), and diethyl ether (≥99.0%) were purchased from Sigma Aldrich. Polyethylene glycol (2000 g mol−1) was purchased from Fluka Chemika. Dimethyl sulfoxide was purchased from J. T. Baker and 1-octanol from Fisher Science Education. Benzene was purchased from TCI. Ethylene oxide gas was purchased from Praxair, condensed into a Schlenk flask, and distilled into a vacuum flask containing calcium hydride. It then was distilled into a round-bottom flask containing a sodium mirror, and finally distilled into a vacuum flask with a stir bar and stored in an explosion proof freezer. Prior to its use in a polymerization, it was again distilled onto a sodium mirror in a graduated ampoule before addition to the reaction vessel. All other solvents and reagents were used as received from Fisher unless otherwise mentioned.Instrumentation
Gel permeation chromatography (GPC) was used to determine the molecular weight and polydispersity of the PEO polymers. A Waters GPC-1, 1515 HPLC Pump and Waters 717Plus Autoinjector were used. The instrument contains Jordi Gel fluorinated DVB columns (1–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 2–10000, 1–500 Å) and a Varian 380-LC Evaporative Light Scattering Detector (ELSD). THF was used as the mobile phase. Empower 1, Waters software, was used to run the samples and analyze the data based on narrow dispersity polystyrene standards purchased from Polymer Laboratories. Proton NMR was obtained on a Bruker DMX 500 MHz High Resolution Nuclear Magnetic Resonance (NMR) Spectrometer with Bruker Topspin 1.3 software. MestReNova software was utilized to analyze the spectra.
000, 2–10000, 1–500 Å) and a Varian 380-LC Evaporative Light Scattering Detector (ELSD). THF was used as the mobile phase. Empower 1, Waters software, was used to run the samples and analyze the data based on narrow dispersity polystyrene standards purchased from Polymer Laboratories. Proton NMR was obtained on a Bruker DMX 500 MHz High Resolution Nuclear Magnetic Resonance (NMR) Spectrometer with Bruker Topspin 1.3 software. MestReNova software was utilized to analyze the spectra.
Methods
Synthesis of macroinitiators
The synthesis of polystyrene endcapped with a hydroxyl group (PS–OH) was performed in a glove box. The solvent, cyclohexane containing approximately 10% by volume benzene, was previously dried over sec-butyllithium and 1,1-diphenylethylene on a vacuum line, and 150 ml was vacuum distilled into a 250 ml vacuum flask and brought into the glove box. In the glove box, 2.56 ml of 1.3 M sec-butyllithium initiator (3.3 mmoles), was then added by syringe to the dry solvent. Next, 11 ml (10 g, 96 mmoles) of styrene, dried over dialkyl magnesium and collected by vacuum distillation, was added and the reaction allowed to stir overnight at room temperature. The color of the solution turned deep orange. Excess ethylene oxide (∼2 ml) was then added and the color of the solution faded. The vacuum flask was removed from the glove box and degassed methanol was added to terminate the reaction. The cloudy solution was allowed to stand overnight. The polymer solution was filtered, followed by precipitation in excess methanol. The resulting solid was dried under vacuum for several days. GPC and 1H NMR were used to characterize the molecular weight and polydispersity of the polymer. PEO homopolymer (2 kg mol−1) and 1-octanol were used as received.Polymerization of PEO: method 1, vacuum distillation
The polymerization of PEO was performed by the following general procedure that varied slightly depending on the starting alcohol. For the hydroxyl endcapped PS, the polymer (3.00 g, 1.15 mmol) was placed in a 500 ml round-bottom vacuum flask equipped with a magnetic stir bar and a high vacuum Teflon valve. To this was added approximately 50 ml of DMSO and 300 ml toluene. Next, 11.5 ml of a 0.100 N KOH in methanol solution (1.15 mmol) was added, taking care to error on the side of too little rather than too much. The reaction flask was then attached to a vacuum line and all subsequent transfers were carried by vacuum distillation on the line. First, the toluene was slowly removed, followed by the addition of approximately 200 ml of degassed THF previously dried over sodium/benzophenone. Next, degassed ethylene oxide, previously dried over CaH2, was distilled into a 250 ml flask containing a sodium mirror, then distilled into a 10 ml graduated cylinder attached to the vacuum line, and from there 1.7 ml (34 mmol) was distilled into the reaction flask.The valve on the vacuum flask was then closed and the flask removed from the line and placed in an oil bath. The reaction mixture was heated to 60 °C for 5–7 days, then quenched with degassed methanol. The polymer solution was filtered and then precipitated by pouring into cold diethyl ether. The solid was collected and placed in a vacuum oven at ambient temperature to dry. GPC and 1H NMR were used to characterize the molecular weight and polydispersity of the PEO polymers. The PEO extension reaction was performed with benzene as an initial solvent instead of toluene. Toluene was used as the initial solvent for all other polymerizations.
Synthesis of PEO polymers: method 2, atmospheric pressure distillation
A second method to synthesize PEO with a Dean–Stark apparatus was used to perform the azeotropic distillation. For a typical synthesis, a 1 L vacuum flask, equipped with a stir bar, approximately 300 ml toluene, 50 ml of DMSO, and an equimolar amount of 0.1 N KOH in methanol and 1-octanol (0.111 ml, 0.7064 mmol) were added. The Dean–Stark apparatus was equipped with a ground-glass joint thermometer and a water-cooled condenser. The vacuum flask was placed in an oil bath on a hot/stir plate and then connected to the Dean–Stark apparatus, leaving the Teflon valve open. The reaction mixture was distilled, with the end point determined by monitoring the temperature of the vapor. Once the water was removed, the Teflon valve was closed as the heat was removed. The reaction flask was allowed to cool to room temperature, fixed to a vacuum line and approximately 200 ml of dry THF was distilled into the flask. Further reaction steps then proceeded as in Method 1.Results and discussion
The equilibrium between alcoholic potassium hydroxide and potassium alkoxide, as shown in Scheme 1, lies far to the left. By Le Chatelier's principle, removal of the water from the right hand side drives the equilibrium towards the alkoxide. Toluene is our solvent of choice, as it forms an azeotrope with both water and methanol, solubilizes a variety of polymers, has a reasonably low boiling temperature, and has fewer health and environmental concerns than benzene. The binary positive azeotrope between methanol and toluene distills at 63.8 °C, with the vapor containing a 0.883 mole fraction of methanol, or 72.4 wt% methanol. The azeotrope with methanol is convenient, as we typically add the KOH by way of a 0.1 N standard solution of KOH in methanol. The positive binary azeotrope between water and toluene distills at 84.1 °C, with the vapor containing a 0.444 mole fraction of water, or 13.5 wt% water.35 The differences between the boiling temperature of the azeotropes and of pure toluene (110.6 °C) are large enough to clearly distinguish them during the distillation.To demonstrate the growth of PEO from a small molecular weight alcohol, we use 1-octanol, an alcohol with sufficiently low vapor pressure to remain in solution while the water is removed by azeotropic distillation with the toluene co-solvent. A PEO polymer with a target MW of 10 kg mol−1 is synthesized in a solution of toluene and THF, both with and without DMSO. The use of DMSO as a co-solvent is studied based on literature reports that it aids in the polymerization of EO.33 During the reaction, the mixture with DMSO turned yellow once the alkoxide had formed, indicating an anion was present. The reaction mixture in the absence of DMSO remained clear.
The PEO homopolymer thus synthesized from 1-octanol is analyzed with 1H NMR and GPC. The polydispersity index (PDI) determined by GPC for the polymer made with DMSO present is 1.21, while without DMSO the PDI is 1.19. The GPC chromatograms of the PEO homopolymers from 1-octanol with and without DMSO are shown in Fig. 1a and b. 1H NMR MW results are shown in Fig. 2. Assigning the singlet at 3.62 ppm to the methylene protons in the backbone of the PEO chain, and the triplet at 0.85 ppm to the terminal methyl group of the 1-octanol, integration of the peak areas gives Mn values of 6.5 kg mol−1 for the polymer grown with DMSO and 10.0 kg mol−1 for the polymer synthesized without DMSO. The lower MW of the PEO synthesized with DMSO corresponds with a lower yield, likely due to EO being less soluble in the reaction mixture containing DMSO. The nearly identical PDI with and without DMSO argues against a termination event. Slower rates of reaction due to lower concentrations of EO dissolved in the reaction mixture are supported by several observations. The first is that the isolated yield of the reaction with DMSO is 77%, and the measured molecular weight is roughly 70% of the target. The second observation is that using a 1:1 ratio of DMSO:THF results in PEO with a MW roughly 10% of the of the target. Changing the ratio to 1:4 DMSO:THF results in PEO with a MW 70% of the target, with all other reaction conditions held constant. Without any DMSO at all, the yield is 99.5%.
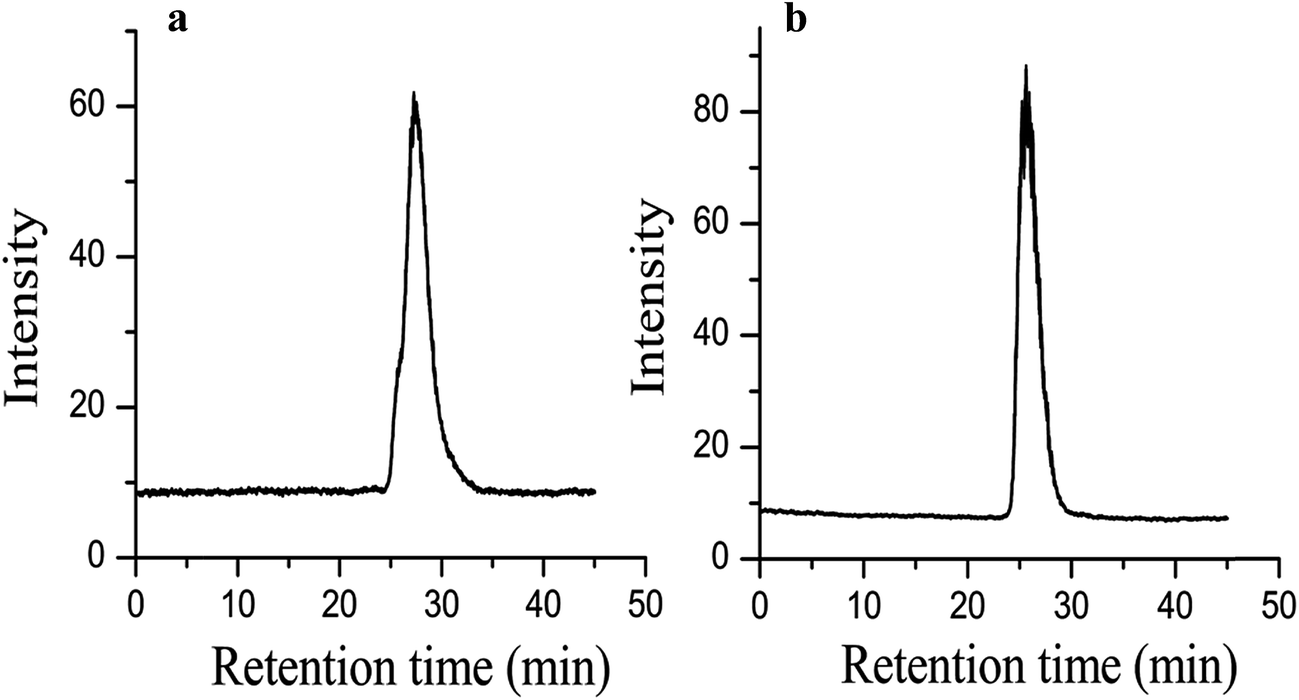 | ||
| Fig. 1 (a) GPC trace of PEO homopolymer from 1-octanol initiator with DMSO (b) GPC trace of PEO homopolymer from 1-octanol without DMSO. | ||
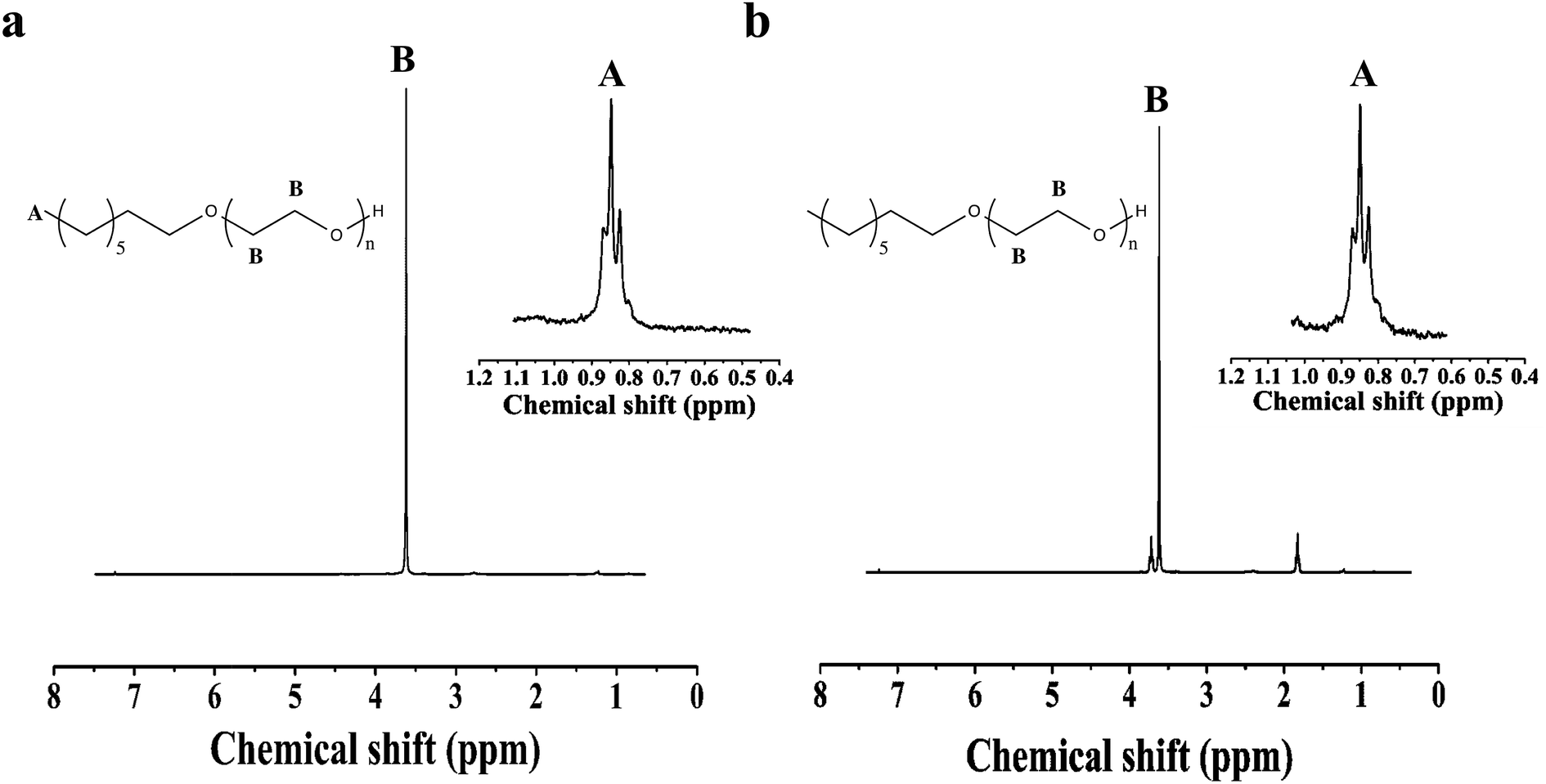 | ||
| Fig. 2 (a) 1H NMR of PEO homopolymer from 1-octanol initiator with DMSO (b) 1H NMR of PEO homopolymer from 1-octanol without DMSO. | ||
1H NMR also provides evidence that the PEO chains are indeed initiated from the alcohol, as the presence of PEO chains not initiated from the alcohol would result in 1H NMR results, which are based on the peak areas associated with the alcohol, being far different than the target MW. The presence of free alcohol is unlikely due to the precipitation of the polymer during workup. Looked at a different way, this means that there was no significant amount of PEO “homopolymer”, or polymer initiated without the intended initiator. As our approach works by removing the water present in the system, no water is available to initiate polymerization and produce homopolymer.
In addition to 1-octanol, propargyl alcohol is used to grow PEO with a target molecular weight of 5 kg mol−1. Due to its thermal sensitivity, method 1, or vacuum distillation, is used to form the alkoxide and no DMSO is present in the system. Once the alkoxide forms, a colorless toluene solution remains. After distilling in dry THF, the solution turns cloudy white prior to heating at 60 °C for 7 days. The PEO synthesized from propargyl alcohol is analyzed with 1H NMR and GPC, giving a Mn value of 6.8 kg mol−1 and a polydispersity index (PDI) of 1.37. Fig. 3 shows the GPC chromatogram and 1H NMR spectrum for the polymer. The chemical shift value at 4.45 ppm corresponds to the methylene group in the initiator. The methylene group in the backbone of the polymer chain is shown as a singlet at 3.60 ppm. Comparing the integration of the peak areas, the calculated Mn from 1H NMR is 6.8 kg mol−1, slightly higher than the target MW, possibly due to not all of the propargyl initiator being converted to the alkoxide. The yield of the reaction is 96%.
 | ||
| Fig. 3 (a) GPC of PEO homopolymer from propargyl alcohol initiator (b) 1H NMR of PEO homopolymer from propargyl alcohol. | ||
Next, block copolymers of PS-b-PEO are synthesized. Hydroxyl terminated polystyrene macroinitiator (PS–OH) is used as the alcohol, and is analyzed by GPC and 1H NMR before using the azeotropic method to grow the PEO block. The PDI by GPC is 1.19, and the Mn is 2.6 kg mol−1 by NMR. From this macroinitiator, PEO with a target MW of 1.3 kg mol−1 is synthesized using method 1. Fig. 4a and b show the overlay of the original polymer (in red) and the block copolymer (in black). The 1H NMR spectrum of the PS–OH macromer is shown in Fig. 4c, with the relevant peaks labeled.
 | ||
| Fig. 4 (a) Overlapping GPC traces of PS–OH and PS-b-PEO with DMSO (b) overlapping GPC traces of PS–OH and PS-b-PEO without DMSO (c) 1H NMR of PS–OH (d) 1H NMR of PS-b-PEO with DMSO (e) 1H NMR of PS-b-PEO without DMSO. | ||
As with the 1-octanol initiated polymerization, the effect of DMSO on the azeotropic initiated PS–OH macromer is investigated. In both cases, with and without DMSO, the yield of the polymerization is better than 98%, and polymers are characterized by GPC and 1H NMR. The GPC chromatogram of the PS-b-PEO with DMSO is shown in Fig. 4a, with an overlaid trace of the PS–OH macromer, and a PDI of 1.21. In comparison, Fig. 4b shows the GPC trace of PS-b-PEO without DMSO, with a PDI of 1.50. The presence of the DMSO appears in this case to give a narrower molecular weight distribution, as seen previously by Quirk et al.33Fig. 4d and e show the 1H NMR spectra of PS-b-PEO with and without DMSO, respectively, each containing the characteristic PEO peak at around 3.6 ppm. The Mn of the PEO block made with DMSO from 1H NMR is 1.1 kg mol−1, close to the target of 1.3 kg mol−1, while the molecular weight of the PEO block without DMSO is 1.9 kg mol−1. It thus appears the effect of the DMSO may be to make the PS–OH macromer a better initiator, possibly by stabilizing the charged chain end outside of the polymer coil, where it is likely buried with more nonpolar solvents.
This method is also used to extend the molecular weight of preformed hydroxyl-terminated PEO polymers. A commercial PEO with a molecular weight of 2.0 kg mol−1 is used with a target of 20 kg mol−1 for the extended polymer. After azeotropic distillation, the reaction mixture turns yellow, suggesting the presence of an anion. Fig. 5a shows overlaid GPC traces of the commercial PEO and the extended PEO chain. The commercial PEO has a PDI of 1.30, and after extension, the PDI decreases to 1.16. Fig. 5b shows the 1H NMR spectrum of the commercial PEO and Fig. 5c is the 1H NMR spectrum of the extended PEO polymer. From Fig. 5c, the molecular weight of the extended PEO is 17.4 kg mol−1, which agrees well with our initial target molecular weight of 20 kg mol−1.
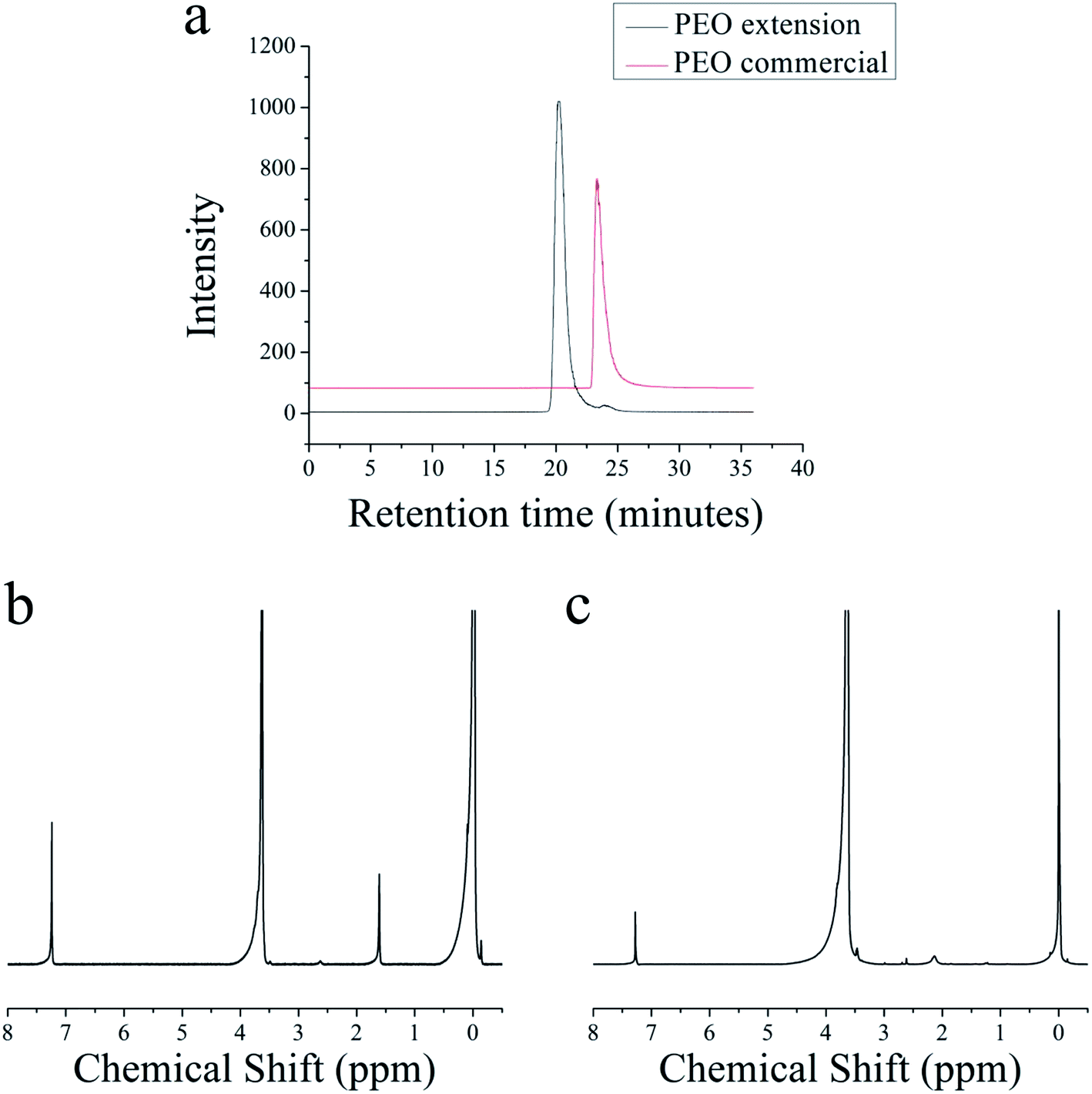 | ||
| Fig. 5 (a) Overlapping GPC traces of commercial PEO before and after extension (b) 1H NMR of commercial PEO (c) 1H NMR of extended PEO chain. | ||
Conclusion
Using azeotropic distillation to remove water and form potassium alkoxides directly from alcohols and KOH, we have demonstrated the synthesis of PEO from small molecular weight alcohols, the formation of PEO block copolymers, and the extension of PEO chains. Table 1 summarizes the polymers synthesized and discussed. Two methods, one under vacuum and the other at atmospheric pressure, were introduced. In addition, the azeotropic approach avoids the often necessary removal of PEO homopolymers and compounds such as naphthalene from the final product. The presence of a co-solvent, DMSO, was also investigated, and it was found to be helpful only for the synthesis of PEO from macromers. This approach to the synthesis of PEO provides an attractive and useful alternative to the current practice of employing air sensitive and difficult to work with alkyl and aryl potassium reagents.| Alcohol | Target Mn (kg mol−1) | M n by 1H NMR (kg mol−1) | MW dispersity (Mw/Mn) | Isolated yield (%) |
|---|---|---|---|---|
| a Dispersity of macroinitiator prior to synthesis of PEO block. | ||||
| 1-Octanol w/DMSO | 10 | 6.5 | 1.21 | 77 |
| 1-Octanol w/o DMSO | 10 | 10.0 | 1.19 | >99 |
| Propargyl alcohol | 5 | 6.8 | 1.37 | 96 |
| PS–OH w/DMSO | 1.3 | 1.1 | 1.21 (1.19)a | >98 |
| PS–OH w/o DMSO | 1.3 | 1.9 | 1.50 (1.19)a | >98 |
| PEO | 20 | 17.4 | 1.16 (1.30)a | 85 |
Acknowledgements
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1247393 and NSF grant CHE-1310453.Notes and references
- N. Ekizoglou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1198–1202 CrossRef CAS
.
- H.-Q. Xie and D. Xie, Prog. Polym. Sci., 1999, 24, 275–313 CrossRef CAS
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, J. Am. Chem. Soc., 2009, 131, 3201–3209 CrossRef CAS PubMed
- M. L. Arnal, V. Balsamo, F. Lopez-Carrasquero, J. Contreras, M. Carrillo, H. Schmalz, V. Abetz, E. Laredo and A. J. Müller, Macromolecules, 2001, 34, 7973–7982 CrossRef CAS
- P. Alexandridis and T. A. Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS
- M. A. Hillmyer and F. S. Bates, Macromolecules, 1996, 29, 6994–7002 CrossRef CAS
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2012, 64, 246–255 CrossRef
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed
- B. K. Money and J. Swenson, Macromolecules, 2013, 46, 6949–6954 CrossRef CAS
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef
- M. J. Barthel, F. H. Schacher and U. S. Schubert, Polym. Chem., 2014, 5, 2647–2662 RSC
- G. Kwon, M. Naito, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, Langmuir, 1993, 9, 945–949 CrossRef CAS
- P. Buskens, M. Wouters, C. Rentrop and Z. Vroon, J. Coat. Technol. Res., 2013, 10, 29–36 CrossRef CAS
-
N. Clinton, P. Matlock, S. D. Cagnon, R. W. Body and V. L. Kylingstad, in Encyclopedia of Polymer Science and Technology, Wiley-Interscience, 1986, pp. 225–322 Search PubMed
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Slomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS
- I. Fallais, J. Devaux and R. Jerome, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1618–1629 CrossRef CAS
- S. Forster and E. Kramer, Macromolecules, 1999, 32, 2783–2785 CrossRef
-
N. Hadjichristidis, S. Pispas and G. Floudas, Block Copolymers: Synthetic Strategies, Physical Properties, and Applications, John Wiley & Sons, Inc., Hoboken, New Jersey, 2003 Search PubMed
- J. Allgaier, A. Poppe, L. Wilner and D. Richter, Macromolecules, 1997, 30, 1582–1586 CrossRef CAS
- Z. Hruska, G. Hurtrez, S. Walter and G. Riess, Polymer, 1992, 33, 2447–2449 CrossRef CAS
- D. Lanson, M. Schappacher, R. Borsali and A. Deffieux, Macromolecules, 2007, 40, 9503–9509 CrossRef CAS
- T. C. Castle, L. R. Hutchings and E. Khosravi, Macromolecules, 2004, 37, 2035–2040 CrossRef CAS
- R. Knischka and P. J. Lutz, Macromolecules, 2000, 33, 315–320 CrossRef CAS
- P. G. Fragouli, H. Iatrou and N. Hadjichristidis, Polymer, 2002, 43, 7141–7144 CrossRef CAS
- N. Ekizoglou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2166–2170 CrossRef CAS
- P. Zhou and H. Xie, Polym. Commun., 1985, 124–130 Search PubMed
- I. Gitsov, A. Simonyan and N. G. Vladimirov, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5136–5148 CrossRef CAS
- T. N. Khan, R. H. Mobbs, C. Price, J. R. Quintana and R. B. Stubbersfield, Eur. Polym. J., 1987, 23, 191–194 CrossRef CAS
- A. Khan and M. Siddiq, J. Appl. Polym. Sci., 2010, 118, 3324–3332 CrossRef CAS
- G. L. Jialanella, E. M. Firer and I. Piirma, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1925–1933 CrossRef CAS
- B. Esswein and M. Moller, Angew. Chem., Int. Ed. Engl., 1996, 35, 623–625 CrossRef CAS
- R. P. Quirk, J. Kim, C. Kausch and M. Chun, Polym. Int., 1996, 39, 3–10 CrossRef CAS
- B. Eßwein, N. Steidl and M. Möller, Macromol. Rapid Commun., 1996, 148, 143–148 CrossRef
-
L. H. Horsley, Azeotropic Data - III, Advances in Chemistry Series 116, American Chemical Society, 1973 Search PubMed
| This journal is © The Royal Society of Chemistry 2016 |