
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUtilization of a new gold/Schiff-base iron(III) complex composite as a highly sensitive voltammetric sensor for determination of epinephrine in the presence of ascorbic acid†
Adam
Gorczyński
,
Maciej
Kubicki
,
Klaudia
Szymkowiak
,
Teresa
Łuczak
* and
Violetta
Patroniak
*
Faculty of Chemistry, Adam Mickiewicz University, Umultowska 89b, 61614 Poznań, Poland. E-mail: violapat@amu.edu.pl; telucz@amu.edu.pl
First published on 19th October 2016
Abstract
The preparation of new materials that can act as systems capable of sensing biologically relevant molecules constitutes a significant modern challenge as well as a necessity oriented towards disease prevention. Subcomponent self-assembly of 2-(methylhydrazino)benzimidazole, 4-tert-butyl-2,6-diformylphenol and Fe(ClO4)2(H2O)6 leads to a new, bimetallic iron(III) complex of the following formula: [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 (1), as established by ESI-MS, FTIR and single crystal X-ray analysis. It is important to note that ligand H3L was also successfully synthesized and characterized for the first time. Compound 1 was successfully deposited on a gold electrode and applied as a voltammetric sensor with respect to epinephrine (EP). Cyclic voltammograms (CVs) proved the catalytic activity of the new, electrochemically prepared composite Au/1 for the oxidation of EP in the presence of ascorbic acid (AA). The respective current peaks were clearly separated from each other, thus enabling selective detection of these compounds coexisting in a mixture. For the prepared sensor a linear relationship between the current response of EP electrooxidation at the potential of peak maximum (ip) and the concentration of EP in solution (cEP) in the presence of constant AA concentration was found in the broad range of cEP (R2 ≥ 0.9997, 1.0 × 10−8 M to 9.0 × 10−4 M) with a high detection limit (7.4 × 10−9 M), excellent reproducibility as well as high stability.
1. Introduction
Epinephrine (adrenaline) (EP) belongs to a class of biogenic aliphatic catecholamines that plays an important role in functioning of the mammalian, hormonal, central nervous and cardiovascular systems.1 In similarity to other hormones, EP exhibits a suppressive effect on the immune system and is thus used as a drug to treat cardiac arrest or dysrhythmias, as a bronchodilator for asthma and for sepsis treatment.2 Development of a simple method for EP detection and determination is an important task from both fundamental and practical points of view in bioscience as well as in medicinal chemistry – especially in neurochemistry – and has resulted in plenty of papers published in the past years. Catecholamines are easily oxidized, thus electrochemical methods appear to be suitable for their quantitative detection.3 However, the final product of epinephrine oxidation – epinephrinechrome – easily transforms into polymer, which makes a serious problem when epinephrine is oxidized directly at the bare electrode due to poisoning and blocking of the electrode surfaces. Another problem is that epinephrine exists in natural environments together with small biomolecules like ascorbic acid, which oxidizes at bare electrodes in almost the same potential region.4,5 It is thus necessary to separate the oxidation potentials of these compounds to enable accurate determination of epinephrine. Among various attempts to overcome the above mentioned problems, much attention has been paid to application of chemically or electrochemically modified electrode surfaces. The most papers have been concerned with the voltammetric behavior of bare carbon, carbon fiber and glassy carbon based electrodes. For example a pyrolytic graphite electrode modified with carbon nanotubes,6–8 a graphite/mesoporous Al-containing SiO2 electrode,9 carbon fiber electrodes covered with overoxidized polypyrrole and DNA10 as well as carbon surface with deposited gold nanoparticles (Au-NPs)11–13 have been tested for selective detection of EP in the presence of interfering compounds. Moreover, glassy carbon electrodes modified with an overoxidized polypyrrole14 or dopamine,15 polycaffeic acid,16 luminal,17 Zn–Al hydroxide,18 5-amino-1,3,4-thiadiazole-2-thiol19 and poly rutin20 films were reported. A glassy carbon electrode covered with self-assembled monolayers (SAMs) of cysteamine21 as well as with osmium and cobalt hexacyanoferrate, hexacyanoruthenate or phtalocyanine complexes8,22–25 have been prepared and investigated upon the electrooxidation of EP. Aside from different carbon materials used as a template for preparing modified electrodes, a promotion of electron transfer in the electrochemical reaction during EP electrooxidation was achieved after application of gold electrodes covered with spontaneously formed SAMs of alkanetiols or other S-containing organic compounds.26–33 It has been proven that the latter electrodes effectively protect the metal surface against adsorption of reaction products that are poisonous for the electrode surface.As a consequence of facile and efficient synthetic protocols, we have recently focused our attention on Schiff-base scaffolded metallosupramolecular architectures, which renders them suitable systems for viable industrial applications.34–36 As a result, novel voltammetric sensor has been developed (Au/[Mn2(H2L)2](ClO4)2 composite, where H3L is ligand – 2,6-bis((2-(1H-benzo[d]imidazol-2-yl)-2-methylhydrazono)methyl)-4-(tert-butyl)phenol) and applied for quantitative detection of dopamine and its interfering biogenic compounds.36 It has struck us that such systems have been recognized as an unexplored niche with regard to deposition onto gold electrodes and sensing of biologically relevant compounds and in turn inspired us to pursue this subject with regard to the other metal ions.37
Despite that a lot of experimental data mentioned above is present, development of novel and easy to synthesize materials capable of simultaneous detection of neurotransmitters in the presence of biogenic molecules, such as AA, is still a challenging task for electrochemists. Motivated by successful application of hardly explored Schiff-base metallosupramolecular architectures as sensors,36 we have prepared new bimetallic Fe(III) complex – [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 (1) (Fig. 1) – successfully deposited it at gold template and evaluated prepared material as electrochemical sensor of EP in the presence of interfering AA.
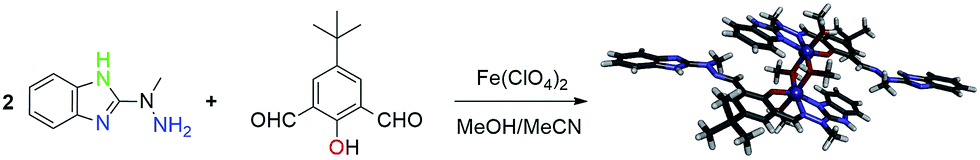 | ||
| Fig. 1 Schematic formation of new Fe(III) bimetallic complex via subcomponent self-assembly approach. | ||
2. Experimental
2.1 Reagents and physical measurements
All chemicals were of analytical grade and used as received without further purification: tetrabutylammonium perchlorate (TBAClO4), lithium perchlorate (LiClO4), NaH2PO4, K2HPO4, NaOH and acetonitrile (ACN) from Merck, epinephrine (EP) from Fluka; L-ascorbic acid (AA) and Fe(ClO4)2(H2O)6 from Sigma; H2SO4, H2O2, EtOHabs, MeOHabs from POCh (Gliwice, Poland). All synthetic manipulations were performed under aerobic conditions using solvents and chemicals as received unless otherwise stated.2.2 Instrumentation
Microanalysis of ligand H3L as well as bimetallic Fe(III) complex 1 was performed using a Perkin-Elmer 2400 CHN microanalyser. IR spectrum of 1 was obtained with a Perkin-Elmer 580 spectrophotometer and peak positions are reported in cm−1. ESI mass spectra of ligand and 1 were determined in methanolic solution with c = ∼10−4 M using a Waters Micromass ZQ spectrometer. The electrochemical measurements were carried out in a conventional three-compartment cell separated by glass frits using an Autolab potentiostat/galvanostat analyzer (AUTOLAB PGSTAT 302N, Eco Chemie, B. V., Utrecht, The Netherlands). The scanning electron microscopy (SEM) analysis was carried out using ZEISS EVO 40 instrument (Germany) with the acceleration voltage of 20 kV. The transmission electron microscopy (TEM) image was obtained using JEM-1200 EX2 (Japan) instrument, operating at 80 kV, after drying the gold colloidal Fe-complex solution covered with the foil strengthened with carbon.2.3 Synthesis of ligand H3L
Synthesis of new Schiff-base ligand H3L was performed under nitrogen to eliminate the detrimental effect of moisture on the condensation efficiency. 2-(1-Methylhydrazinyl)-1H-benzo[d]imidazole (56.0 mg, 0.347 mmol)36 and 4-tert-butyl-2,6-diformylphenol (43.2 mg, 0.17 mmol) were transferred to two-necked round bottom flask equipped with the reflux condenser that was previously flushed with inert gas. 10 ml of absolute ethanol was added via syringe and the mixture was stirred on magnetic stirrer, resulting in formation of transparent yellow solution. The mixture was heated for a few hours under reflux what followed precipitation of yellow solid. The content was cooled to room temperature, left to stir overnight and the precipitate was subsequently filtered and washed well with ethyl alcohol and diethyl ether respectively. Recrystallization from DMSO/water system resulted in microcrystalline yellow solid that was dried under vacuum in 50 °C. Yield: 73.15 mg (87.0%).
1H NMR (500 MHz, d6-DMSO): δ = 1.42 (s, 9H), 3.71 (s, 6H), 7.00–7.06 (m, 4H), 7.35–7.37 (m, 4H), 7.90 (s, 2H), 8.21 (s, 2H), 10.53 (s, 1H), 11.79 (s, 2H) ppm. 13C NMR (125 MHz, d6-DMSO): δ = 31.4, 31.8, 34.1, 109.9, 116.1, 119.7, 120.4, 120.7, 125.6, 134.1, 136.5, 142.0, 142.6, 151.8, 153.4 ppm; elemental analysis calc. for [C28H30N8O] (494.59): C, 68.00; H, 6.11; N, 22.66; found: C, 66.09; H, 6.21; N, 22.61%. ESI-MS(+) m/z: 495 [(H4L)]+, 517 [H3L + Na+]+, 533 [H3L + K+]+. Selected IR (KBr, cm−1): ν(O–H) 3637(w), ν(N–H) 3500–2200 (max. at 3158) (m br); ν(C–H)ar 3085(m), 3055(m); νas(CH3) 2962(m), νas(CH3) 2929(m); νs(CH3) 2905(w), 2868(w); ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)ar, ν(CN) 1626(vs.), 1597(vs.), 1561(vs.), 1480 (s), 1461(s), 1448(s), 1414(m), 1385(m), 1353(w), 1317(m), 1280(m), 1224(m), ν(C–O) 1264(m); γ(C–H)ar, ρ(C–H)ar 1211(s), 1156(m), 1044(m), 1005(m), 945(s), 821(w), 743(s), 699(w), 677(w), 533(w), 433(w).
C)ar, ν(CN) 1626(vs.), 1597(vs.), 1561(vs.), 1480 (s), 1461(s), 1448(s), 1414(m), 1385(m), 1353(w), 1317(m), 1280(m), 1224(m), ν(C–O) 1264(m); γ(C–H)ar, ρ(C–H)ar 1211(s), 1156(m), 1044(m), 1005(m), 945(s), 821(w), 743(s), 699(w), 677(w), 533(w), 433(w).
Crystals suitable for X-ray diffraction were obtained from acidified solution of MeOH/toluene at lowered temperature.
2.4 Synthesis of iron(III) complex
Bimetallic iron(III) complex 1 was synthesized in two different ways: method A (similar manner to the recently published one-pot template reaction36 with some notable changes), method B (direct complexation with ligand H3L).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution of Fe(ClO4)2(H2O)6 (58.8 mg, 0.162 mmol), what resulted in formation of black solution. Such system was stirred for 48 h at room temperature without sealing, providing direct access of air. The reaction mixture was subsequently concentrated in vacuo, precipitated with diethyl ether, and recrystallized from MeOH/Et2O at lowered temperature. Single crystal suitable for X-ray analysis was obtained by means of vial-to-vial diffusion at 4 °C from MeOH/iPr2O system. (cf. crystallographic data). [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 (1): yield: 85.4 mg, 61.1%. Elemental analysis calc. for [C60H86Cl4Fe2N16O28] (1) (1624.85): C, 44.35; H, 4.59; N, 13.79; found: C, 44.13; H, 4.73; N, 13.73%. ESI-MS(+) m/z (%): 548 (100) [Fe(HL)]+, 1095 (10) [Fe2(HL)2–H+]+, 1127 (<5) [Fe2(HL)2–H+ + (MeOH)]+, 1195 (<5) {[Fe2(HL)2](ClO4)}+. Selected IR (KBr, cm−1): ν(O–H) 3700–2200 (m, br), ν(O–H) 3638(w), ν(N–H) 3352(m); ν(C–H)ar 3095(w), 3066(w), 3015 (w); νas(CH3) 2962(m), νas(CH3) 2952(m); νs(CH3) 2905(w), 2867(w); ν(CC)ar, ν(CN) 1659(vs.), 1610(vs.), 1592(vs.), 1556(m), 1546(m), 1494(m), 1474 (s), 1467(s), 1449(s), 1394(m), 1364(s), 1331(w), 1288(m), 1232(m), 1220(m), ν(C–O) 1257(m); δ(OClO) 1146(vs.), 1121(vs.), 1108(vs.), 1087(vs.), γ(C–H)ar, ρ(C–H)ar 1052(s), 1008(m), 958(m), 929(m), 826(m), 745(m), 636(s), 529(w), 501(w); νas(FeOFe) 855(w), γ(ClO) 626(s).
:1 v/v) solution of Fe(ClO4)2(H2O)6 (14.9 mg, 0.041 mmol) was added what resulted in formation of black solution. Such system was stirred for 48 h at room temperature without sealing, providing direct access of air. Work-up was identical to the one presented in method A. Formed product was determined to be identical to the one formed by the template approach.
:1 v/v) solution of Fe(ClO4)2(H2O)6 (58.8 mg, 0.162 mmol), what resulted in formation of black solution. Such system was stirred for 48 h at room temperature without sealing, providing direct access of air. The reaction mixture was subsequently concentrated in vacuo, precipitated with diethyl ether, and recrystallized from MeOH/Et2O at lowered temperature. Single crystal suitable for X-ray analysis was obtained by means of vial-to-vial diffusion at 4 °C from MeOH/iPr2O system. (cf. crystallographic data). [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 (1): yield: 85.4 mg, 61.1%. Elemental analysis calc. for [C60H86Cl4Fe2N16O28] (1) (1624.85): C, 44.35; H, 4.59; N, 13.79; found: C, 44.13; H, 4.73; N, 13.73%. ESI-MS(+) m/z (%): 548 (100) [Fe(HL)]+, 1095 (10) [Fe2(HL)2–H+]+, 1127 (<5) [Fe2(HL)2–H+ + (MeOH)]+, 1195 (<5) {[Fe2(HL)2](ClO4)}+. Selected IR (KBr, cm−1): ν(O–H) 3700–2200 (m, br), ν(O–H) 3638(w), ν(N–H) 3352(m); ν(C–H)ar 3095(w), 3066(w), 3015 (w); νas(CH3) 2962(m), νas(CH3) 2952(m); νs(CH3) 2905(w), 2867(w); ν(CC)ar, ν(CN) 1659(vs.), 1610(vs.), 1592(vs.), 1556(m), 1546(m), 1494(m), 1474 (s), 1467(s), 1449(s), 1394(m), 1364(s), 1331(w), 1288(m), 1232(m), 1220(m), ν(C–O) 1257(m); δ(OClO) 1146(vs.), 1121(vs.), 1108(vs.), 1087(vs.), γ(C–H)ar, ρ(C–H)ar 1052(s), 1008(m), 958(m), 929(m), 826(m), 745(m), 636(s), 529(w), 501(w); νas(FeOFe) 855(w), γ(ClO) 626(s).
:1 v/v) solution of Fe(ClO4)2(H2O)6 (14.9 mg, 0.041 mmol) was added what resulted in formation of black solution. Such system was stirred for 48 h at room temperature without sealing, providing direct access of air. Work-up was identical to the one presented in method A. Formed product was determined to be identical to the one formed by the template approach.
2.5 X-ray crystallography
Diffraction data were collected at 100(1) K by the ω-scan technique on Agilent Technologies XCalibur four-circle diffractometer with Eos CCD detector, equipped with Nova microfocus MoKα radiation source (λ = 0.71073 Å) for 1, and for [H5L](ClO4)2 at room temperature on Agilent Technologies SuperNova four-circle diffractometer with Atlas CCD detector and mirror-monochromated CuKα radiation (λ = 1.54178 Å). The data were corrected for Lorentz-polarization as well as for absorption effects.38 Precise unit-cell parameters were determined by a least-squares fit of reflections of the highest intensity, chosen from the whole experiment. The structures were solved with SIR92 (ref. 39) and refined with the full-matrix least-squares procedure on F2 by SHELXL-2013.40 The structure of 1 contains voids filled with diffused electron density therefore the SQUEEZE procedure41 was applied. All non-hydrogen atoms were refined anisotropically, hydrogen atoms were placed in idealized positions and refined as ‘riding model’ with isotropic displacement parameters set at 1.2 (1.5 for methyl or hydroxyl groups) times Ueq of appropriate carrier atoms. The crystals of 1 (maybe due to the disordered solvent) were of relatively poor quality, and in consequence weak restraints were applied to the displacement parameters of some atoms. The crystals of protonated ligand turned out to be twinned, (which was taken into account during both data reduction and refinement); the BASF factor, describing the content of one of the component41 was refined at 79.0(3)%.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.6236(17) Å, b = 13.530(2) Å, c = 16.128(2) Å, α = 66.459(15)°, β = 87.591(12)°, γ = 78.066(14)°, V = 2077.2(6) Å3, Z = 1, F(000) = 842, dx = 1.308 g cm−3, μ = 0.55 mm−1. 15018 reflections measured, 7301 symmetry-independent (Rint = 13.94%), 2549 with I > 2σ(I). Final R[I > 2σ(I)] = 9.86%, wR2[I > 2σ(I)] = 15.47%, R[all reflections] = 20.02%, wR2[all reflections] = 18.33%, S = 0.93, max/min Δρ = 0.80/−0.40 e Å−3.
, a = 10.6236(17) Å, b = 13.530(2) Å, c = 16.128(2) Å, α = 66.459(15)°, β = 87.591(12)°, γ = 78.066(14)°, V = 2077.2(6) Å3, Z = 1, F(000) = 842, dx = 1.308 g cm−3, μ = 0.55 mm−1. 15018 reflections measured, 7301 symmetry-independent (Rint = 13.94%), 2549 with I > 2σ(I). Final R[I > 2σ(I)] = 9.86%, wR2[I > 2σ(I)] = 15.47%, R[all reflections] = 20.02%, wR2[all reflections] = 18.33%, S = 0.93, max/min Δρ = 0.80/−0.40 e Å−3.
[H5L](ClO4)2–2(C28H32N8O)2+·4(ClO4)−·C7H8·CH3OH, Mr = 1515.21, triclinic, P, a = 11.4426(5) Å, b = 13.0011(5) Å, c = 24.5126(10) Å, α = 90.991(3)°, β = 91.953(4)°, γ = 92.005(4)°, V = 3641.7(3) Å3, Z = 2, dx = 1.38 g cm−3, μ = 2.16 mm−1, F(000) = 1584. 25627 reflections collected up to 2θ = 67.5°, 12871 symmetry independent, (Rint = 8.60%), 7644 with I > 2σ(I). Final R[I > 2σ(I)] = 10.20%, wR2[I > 2σ(I)] = 23.13%, R[all refl.] = 14.69%, wR2[all refl.] = 26.03%, S = 1.05, max/min Δρ = 1.52/−0.58 e Å−3.
Crystallographic data (excluding structure factors) for the structural analysis has been deposited with the Cambridge Crystallographic Data Centre, no. CCDC-1407322 (1) and CCDC-1482453 ([H5L](ClO4)2).†
2.6 Electrode activation and modification
All solutions under investigation were prepared prior to each experiment, using Millipore Milli-Q-water or acetonitrile (ACN) 0.1 M TBAClO4, 0.1 M LiClO4 or phosphate buffer (0.6 M pH = 7) served as a supporting electrolyte solution. All solutions were deaerated by argon (99.998%) before measurements performed at room temperature.The working electrode was either the bare gold electrode (99.999%, Polish State Mint) or the gold electrode modified with the Fe-complex 1 of 0.03 cm2 geometric area. A gold sheet was used as an auxiliary electrode (10 cm2), whereas the saturated calomel electrode (SCE) and the Ag/Ag+ in ACN electrodes were used as reference electrode in aqueous and ACN solution, respectively.
Before any experiment, the bare gold electrode was polished with aluminium slurries of successively decreasing final grades down to 0.05 μm (Buehler) on polishing cloths (Buehler). Next, the polished electrode was rinsed carefully with acetone and water, respectively. After that the electrode was electrochemically activated by cycling in the potential range between E = −0.7 V and E = 0.6 V vs. SCE in the phosphate buffer (0.06 M, pH = 7), with the potential scan rate, dE/dt = 0.1 V s−1 until a stable cyclic voltammogram (acronyms CV or i–E curve) was obtained. This procedure avoids structural changes on the gold surface.42
In order to obtain a gold electrode modified with SAM made of 1, the activated Au template was electrochemically cycled in 0.1 M TBAClO4 supporting electrolyte solution in ACN with addition of 0.001 M Fe-complex at dE/dt = 0.1 V s−1, until a stable CV was obtained. The electrochemical properties of the modified electrode were checked over two weeks and were found to be stable. The consecutive series of voltammetric experiments performed each day were separated by rest periods at open circuit in without solution. Before re-use in electrochemical measurements electrode was washed with acetone and dried in an argon stream. The self-assembled layers can be removed from the modified Au surface by dipping it (for about 5 minutes) in a Piranha solution, i.e. in a mixture 3:1 of aqueous solutions of 96% H2SO4 and 30% H2O2. In order to recover the proposed sensors, the above described procedure of polishing, activation and modification of the bare Au electrodes must be repeated.
3. Results and discussion
Subcomponent self-assembly approach43 allowed us to synthesize new, bimetallic iron(III) complex, metal ions bound within N′N′O′N′N Schiff-base scaffold H3L in unprecedented manner (cf. Section 3.1 and Fig. 1). During the course of the reaction, native Fe(II) metal ions are oxidized by air to Fe(III) species and coordinate with in situ formed ligand H3L to form [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 (1). Electrospray ionization mass spectrometry, elementary analysis as well as FT-IR spectroscopy confirmed formation of FeOFeO bridged bimetallic species, whereas its structure in the solid state has been unambiguously confirmed via single X-ray crystallography. Despite the fact that the major species found under ESI-MS studies are monometallic [Fe(HL)]+ positive ions, we believe the solid state structure is retained in solution on the basis of the following premises: (i) it is still possible to find signals from bimetallic species [Fe2(HL)2–H+]+, [Fe2(HL)2(MeO)]+, {[Fe2(HL)2](ClO4)}+ (ii) all observed systems are amenable to the applied cone-voltage. It is also of notice that observed μ-oxo Fe2O2 motif is of relevance with regard to nonheme iron-containing enzymes44–46 and quite unexpectedly herein, it is formed via coordination with adventitious methoxy groups rather than with exploitation of the phenoxy moieties of H3L ligand. The latter situation was observed in [Mn2(H2L)2](ClO4)2 manganese(II) complex, which we have recently demonstrated to form electrochemical sensor at the Au template with regard to sensing of dopamine in the presence of interfering biogenic compounds.36 Herein however, we also demonstrated that the template approach is not the only one that leads to the desired product. It was possible to isolate the new Schiff-base ligand and subdue it to the full spectroscopic characterization. Furthermore, it was possible to transform it into a diprotonated cationic salt [H5L](ClO4)2 and unambiguously confirm its structure in the solid state (vide infra). We were able to complexate H3L with iron(II) salt and the final product turned out to be identical with the one obtained via the subcomponent self-assembly approach.3.1 Crystallographic data
Both systems that comprise the H3L Schiff-base ligand have been crystallized and characterized in the solid state, either in the form of dicationic salt [H5L]2+ or bimetallic Fe(III) complex (1).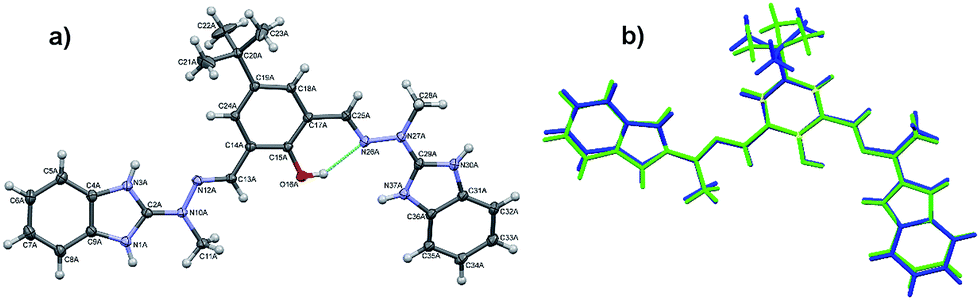 | ||
| Fig. 2 Anisotropic ellipsoid representation of the molecule A of a [H5L]2+ cation (a); ellipsoids are drawn at the 50% probability level, hydrogen atoms are shown as spheres of arbitrary radii, hydrogen bond is shown as thin blue line; comparison of two symmetry-independent cations (fitting of the central rings – b). | ||
The cations are almost planar, the dihedral angles between the mean planes of the terminal ring systems are 13.5° and 10.2° (the relevant geometrical features of the ligands are listed in the Table S1, ESI†). Intramolecular OH⋯N hydrogen bonds help in adopting such conformation. In the crystal structure, besides four perchlorate anions, methanol and (heavily disordered) toluene molecules were found. The crystal structure results – apart from the electrostatic interactions between charged species – from the extensive network of hydrogen bonds, involving all strong hydrogen bond donors (Fig. 3). The details of the hydrogen bonds are listed in Table S2, ESI.†
 | ||
| Fig. 3 A crystal structure of [H5L](ClO4)2 cation as seen along b-direction. Anions and solvent molecules are shown in van der Waals spheres representation in order to visualize their filling-structure role. | ||
(Fig. 4).
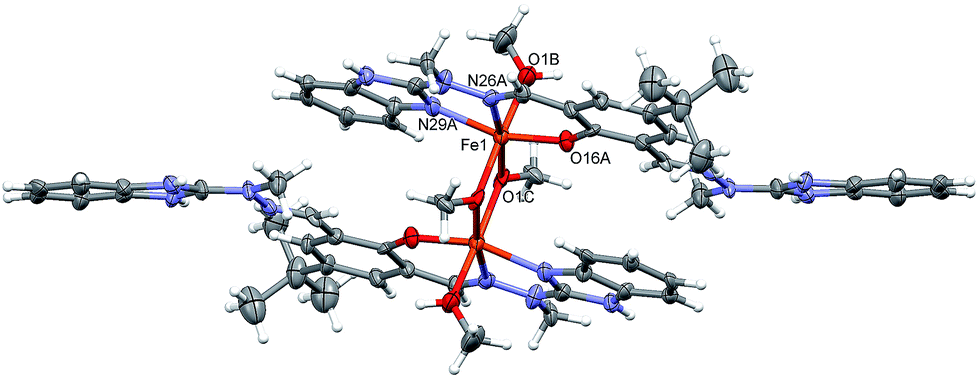 | ||
| Fig. 4 Anisotropic ellipsoid representation of the cation; ellipsoids are drawn at the 30% probability level, hydrogen atoms are shown as spheres of arbitrary radii. The primed atoms are related with the unprimed ones by symmetry operation 1 − x, 1 − y, 1 − z. | ||
Table 1 lists the relevant geometrical parameters. Fe atoms are six-coordinated, by three atoms (NNO) from H3L molecule: two oxygen atoms from bridging methoxy anion, and one oxygen atom from additional methanol. The coordination is closed to octahedral (cf.Table 1), and the central FeOFeO ring is planar. This was also supported by SHAPE measurement calculations,47,48 which indicate the level of deviation from the perfect polyhedron (SO) by means of continuous shape measurements (CShM). Indeed, the lowest value was encountered for octahedron (S = 2.04), showing that the site symmetry is best described as Oh. We did also compare the symmetry of the previously synthesized [Mn2(H2L)2](ClO4)2 complex (Table S3, ESI†) and concluded that the trigonal prism geometry of Mn(II) cations (D3h site symmetry) might indirectly confer to different conformation of ligand H3L and thus different chemisorption onto the Au electrode (Fig. S1 and S2†). The ligands are slightly twisted dihedral angles between mean planes of the terminal ring systems are 18.3(2)°.
| Bond length [Å] | Bond angle [°] | ||
|---|---|---|---|
| Fe1–O16A | 1.879(16) | O16A-Fe1-N29A | 157.1(3) |
| Fe1–N29A | 2.052(7) | O1C–Fe1–N26A | 168.8(3) |
| Fe1–N26A | 2.185(8) | O1B–Fe1–O1Ci | 169.5(3) |
| Fe1–O1C | 1.957(6) | Fe1–O1C–Fe1′ | 103.0(3) |
| Fe1–O1B | 2.101(7) | — | — |
| Fe1–O1Ci | 2.002(7) | — | — |
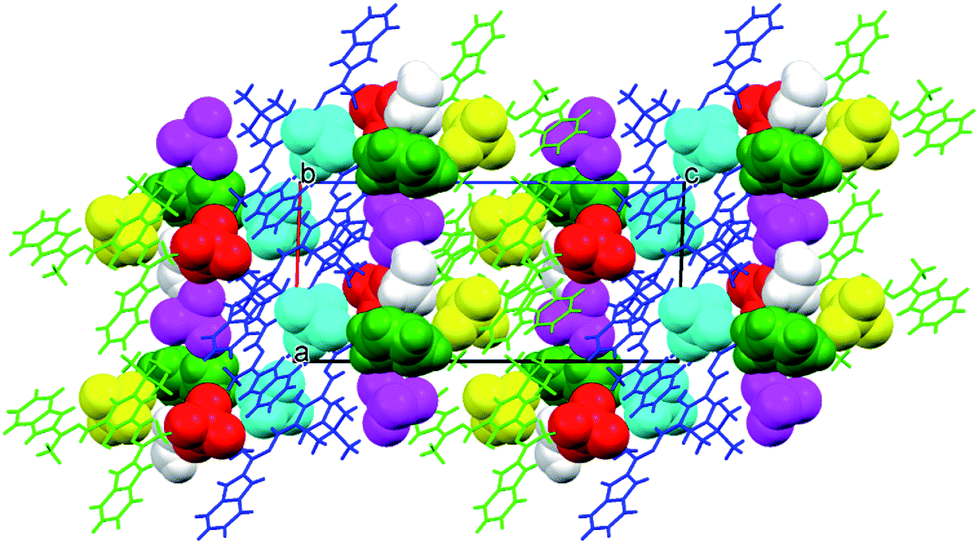
In the crystal structure there are voids which are filled by some electron density, interpreted as disordered water molecules. Hydrogen bonds (N–H⋯O and O–H⋯O) connect the complex with perchlorate anions, and – together with the electrostatic interactions between charged species – might be regarded as a driving force for the crystal structure (Fig. 5).
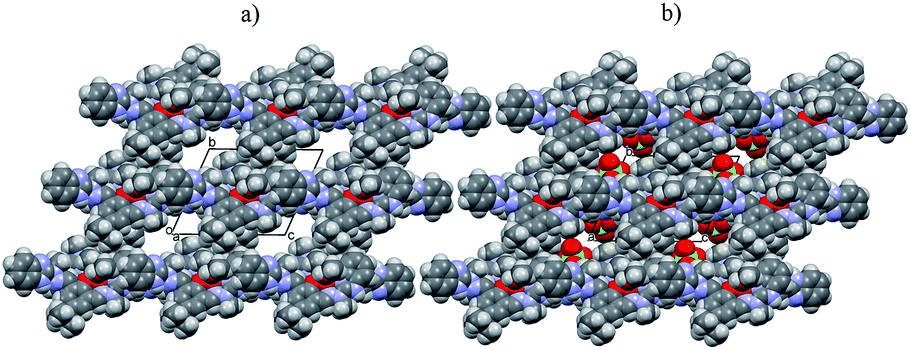 | ||
| Fig. 5 Crystal lattice of [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex as seen along a direction; voids present within the crystal lattice are filled by perchlorate counterions (as shown in (b) and omitted in (a)). | ||
3.2 Electrooxidation of prepared complex on a gold electrode and its characterization
The cyclic voltammograms of the bare polycrystalline gold electrode recorded in 0.1 M tetrabutyloammonium perchlorate (TBAClO4) in acetonitrile (ACN) as a supporting electrolyte solution is presented on Fig. 6a (curve 1). CV obtained in 0.1 M LiClO4 in ACN solution is attached to Fig. 6a (curve 2) for comparative issues.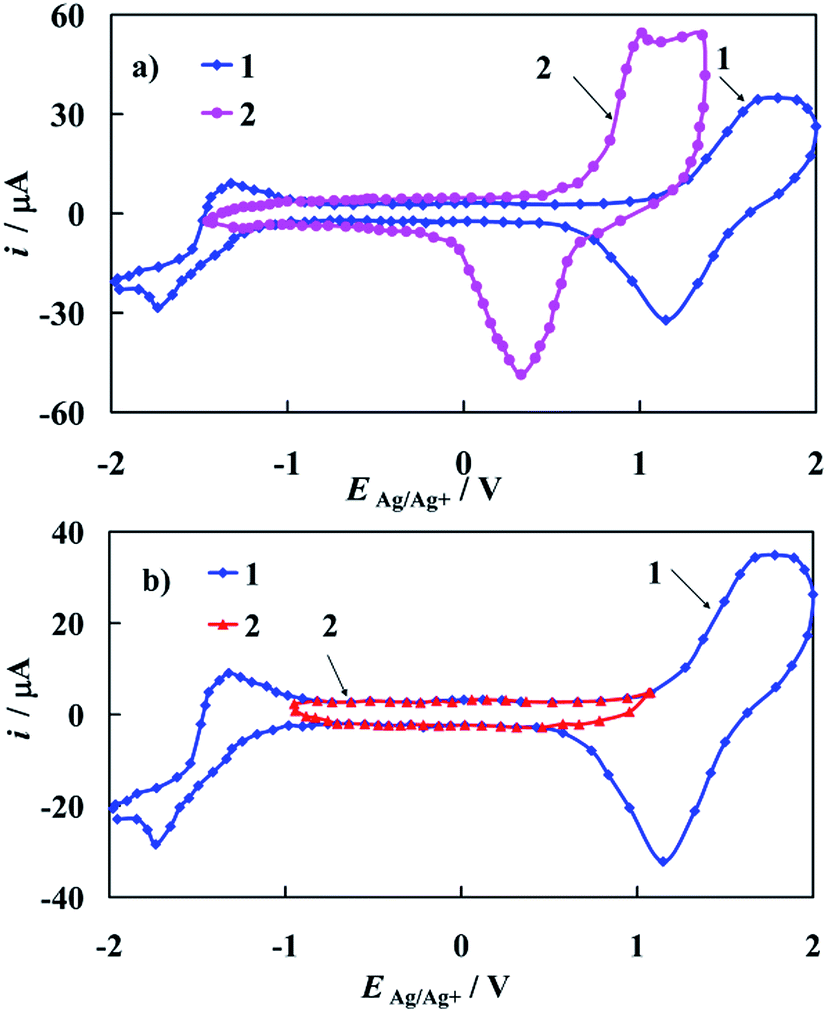 | ||
| Fig. 6 (a) Cyclic voltammograms of polycrystalline gold electrode recorded in: (1) 0.1 M TBAClO4 and (2) in 0.1 M LiClO4 ACN solution. (b) (1) The same solution as in 1(a) with (2) depicting the potential range of the electrical double layer of gold electrode. dE/dt = 0.1 V s−1. | ||
A pair of anodic/cathodic peaks, observed at curve 1 in Fig. 6a, at 1.65 V vs. Ag/Ag+ in the anodic scan and at 1.15 V vs. Ag/Ag+ in the cathodic scan respectively is responsible for oxidation and reduction of a gold oxide layer. Moreover, a couple of peaks visible at −1.75 V and −1.35 V vs. Ag/Ag+ is assigned to the electrochemical behavior of TBA cation. The last conclusion is very important from the electrochemical point of view, since in certain papers that concern complexes studied in the same supporting electrolyte (ACN), the latter pair of peaks was incorrectly considered as coming from the studied complex instead of TBA cation. It is also worth noting that this pair of peaks should be not assigned to the ClO4− anion. This is confirmed by the shape of curve 2 presented in Fig. 6a. Curves 1 and 2 differ considerably despite being recorded in the solutions of supporting electrolyte with different cations. A characteristic feature of the bare gold electrode (Fig. 6b, curve 1) is a wide potential region where no faradic reactions occur (Fig. 6b, curve 2). In this potential region only a charging and discharging of the electrical double layer of the gold electrode occurs (Fig. 6b, curve 2), therefore other electrochemical processes can be studied in this electrode potential window.
The representative set of cyclic voltammograms recorded at the gold electrode in the supporting electrolyte solution and in the presence of the increasing concentration of [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex is depicted in Fig. 7.
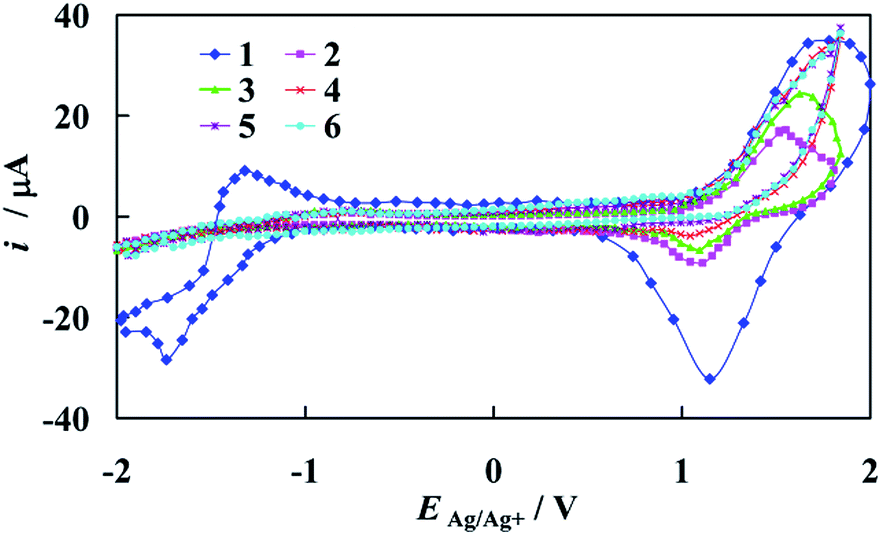 | ||
| Fig. 7 Cyclic voltammograms of polycrystalline gold electrode recorded in: (1) a supporting electrolyte solution: 0.1 M TBAClO4 in ACN and with addition of (2): 1 μM; (3) 10 μM and (4) 50 μM; (5) 100 μM; (6) 1000 μM of the [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4. dE/dt = 0.1 V s−1. | ||
As can be seen, a successive increase of the amount of Fe-complex in solution results in strong adsorption and blocking of the gold surface in the potential range of gold oxide oxidation/reduction. The range of the electric double layer almost does not change and, again the electrode surface is covered by the studied complex in the region of TBA cation. It should be noted that on the recorded i–E curves, peaks which would demonstrate the presence of Fe(III) in the used complex molecules are not present. This stands in contrast with previously presented Mn-complex within the same Schiff base scaffold H3L as dopamine sensor,36 but becomes understandable when comparison of their structures is made.
Formation of self-assembled monolayers (SAMs) from metallosupramolecular architectures on the solid surfaces is dependent on: (i) information encoded within organic ligand and chosen metal ion that affect the final structure of the complex; (ii) nature of the substrate (template) that influences the adsorption behavior of the system under study i.e. the electrocatalyst. Presented bicompartmental Schiff-base ligand has the potential to form various metal ion complexes, in accordance to the exhibited conformation (Fig. 8).
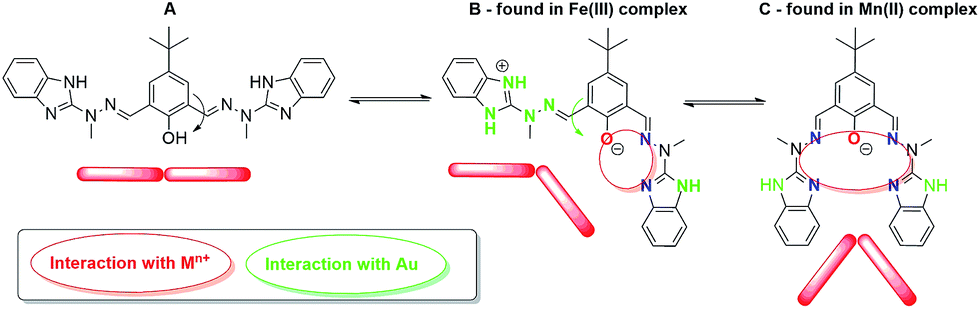 | ||
| Fig. 8 Conformation changes of ligand H3L that outcome the final structure of [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex; A – transoid–transoid, B – transoid–cisoid, C – cisoid–cisoid. | ||
H3L ligand was found to exhibit conformation C in the recently published36 electrocatalyst [Mn2(H2L)2](ClO4)2, whereas herein presented [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex exhibits conformation B. Bearing in mind that both Mn(II) and Fe(III) ions are isoelectronic, such situation is unprecedented and is of prime importance, thus resulting in two important implications. Firstly, we have established that previously formed conformation C that results in shielded, μ-phenoxo bridged bimetallic complexes is not mandatory to form stable composites at gold electrode, since surface coverage of 1 at Au template has been achieved herein (no hydrolysis of iron was observed) as well as is permanent under ambient conditions (vide infra). Secondly, the mode of adsorption may be anticipated to differ, since relative orientation of the NH groups from benzimidazole moieties in iron and manganese electrocatalysts varies. Please also note that despite being isoelectronic, the central metal ions differ in the symmetry as revealed by SHAPE calculations (Section 3.1.). As a result, planar chemisorption of complex 1 is anticipated, contrary to pendant-type chemisorption in [Mn2(H2L)2](ClO4)2i.e. perpendicular with respect to the orientation of Mn(II)–Mn(II) molecular axis. (ESI – Fig. S1†) Such situation would result in ca. 9 times lower concentration of Fe-complex than Mn-complex at the surface of the electrode. This would be the reason that ratio of the Fe concentration to the concentration of all complex in the supporting electrolyte solution is very small, firmly too low, to give peak which may be assigned to the Fe(III) ion.
By taking into account the crystallographic structure of the studied [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex, it is evident that it can be considered as a good candidate to form SAMs on the bare gold surface. It is commonly known that gold is characterized by a good affinity to nitrogen.49,50 It is generally accepted that chemisorption of modified functionalized compounds on gold is preceded by the oxidative abstraction of hydrogen in our case from N–H bonds, whereas the “rest” of modified molecules create an ordered structure at the electrode turned at a tilt angle towards the electrolyte solution.51 The firm attachment of Fe-complex to the gold electrode surface (Au) herein is realized via the covalent bonds between the Au and N atoms from Schiff base ligand studied complex molecule as well as π-stacking interactions. (ESI – Fig. S2†) This has been also confirmed by SEM images of the formed [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4/Au composite (ESI – Fig. S3†).
A strong adsorption at the gold electrode surface was confirmed in separate experiments in which the electrode was cycled in solution with the Fe-complex, and then rinsed copiously with ACN, drying and dipped in the fresh supporting electrolyte. The resulting voltammogram was the same as before electrode washing. Such an experiment, with the same electrode has been repeated each day during two weeks and the recorded i–E curve did not change in shape as compared to that obtained at the beginning of the presented project. We also managed to isolate and characterize the new Schiff-base ligand H3L to check if the deposition of the sole ligand would be possible on the bare Au electrode. Remarkably, the electrochemically mediated deposition of ligand does not occur, what is another proof that formation of SAMs – here in the form of [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4/Au composite material – is possible, provided that H3L was complexated with metallic centers.
3.3 Oxidation of epinephrine at bare and modified gold electrodes
From literature it is well know that formation of self-assembled monolayers (SAMs) are one of the means for functionalization of various electrodes. They are used in the cases when investigation of some compounds on unmodified electrodes disappoints or becomes troublesome. For the above described reasons, such a case is an immanent trait during epinephrine electrooxidation on the bare gold electrode.Electrochemical oxidation of epinephrine goes through three steps, according to the ECE mechanism (where: E and C denotes the electrochemical and the chemical step, respectively) presented below in Fig. 9.3
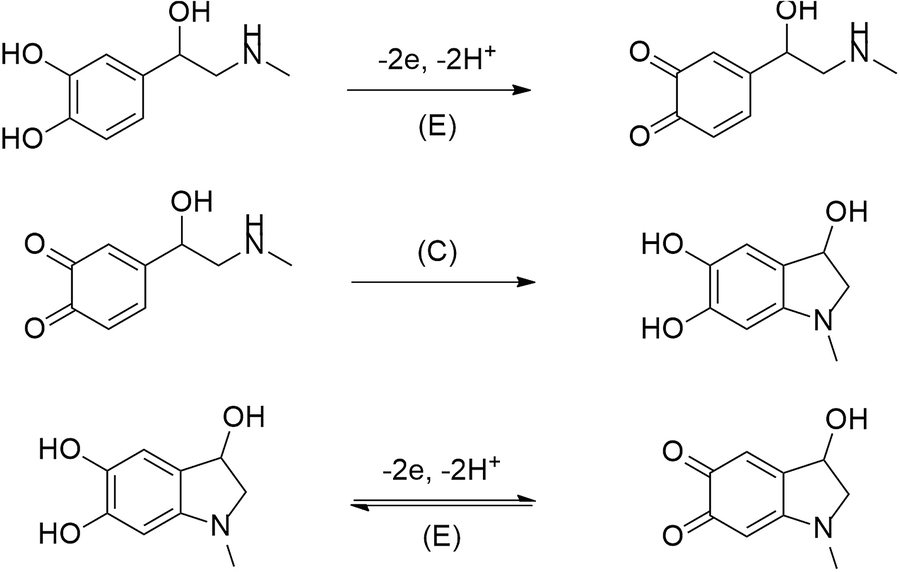 | ||
| Fig. 9 Electrooxidation of epinephrine on electrodes. | ||
Fig. 10a shows the stable voltammetric response (after the 5th scan) of the bare gold electrode in the presence of EP. The epinephrine oxidation at the bare gold electrode proceeds in two potential ranges. The anodic peak with a maximum at E = 0.5 V vs. Ag/Ag+ corresponds to irreversible electrooxidation of EP to o-epinephrinequinone after exchange of 2 electrons and 2 protons. Epinephrinequinone contains both an electron-deficient ring and an electron-donating amine group. When the latter group becomes deprotonated, the molecule can undergo a 1,4 Michael addition, which results in a cyclization reaction.48 Therefore, in the next step o-epinephrinequinone transforms into leucoepinephrinechrome via a chemical intramolecular 1,4-Michael addition.3,50 A subsequent couple of peaks at 0.05 V vs. Ag/Ag+ and 0.11 V vs. Ag/Ag+, appearing during the following cathodic and anodic scans, is related to the reversible electrooxidation of leucoepinephrinechrome to epinephrinechrome and its regeneration after reversal of the potential scan direction.
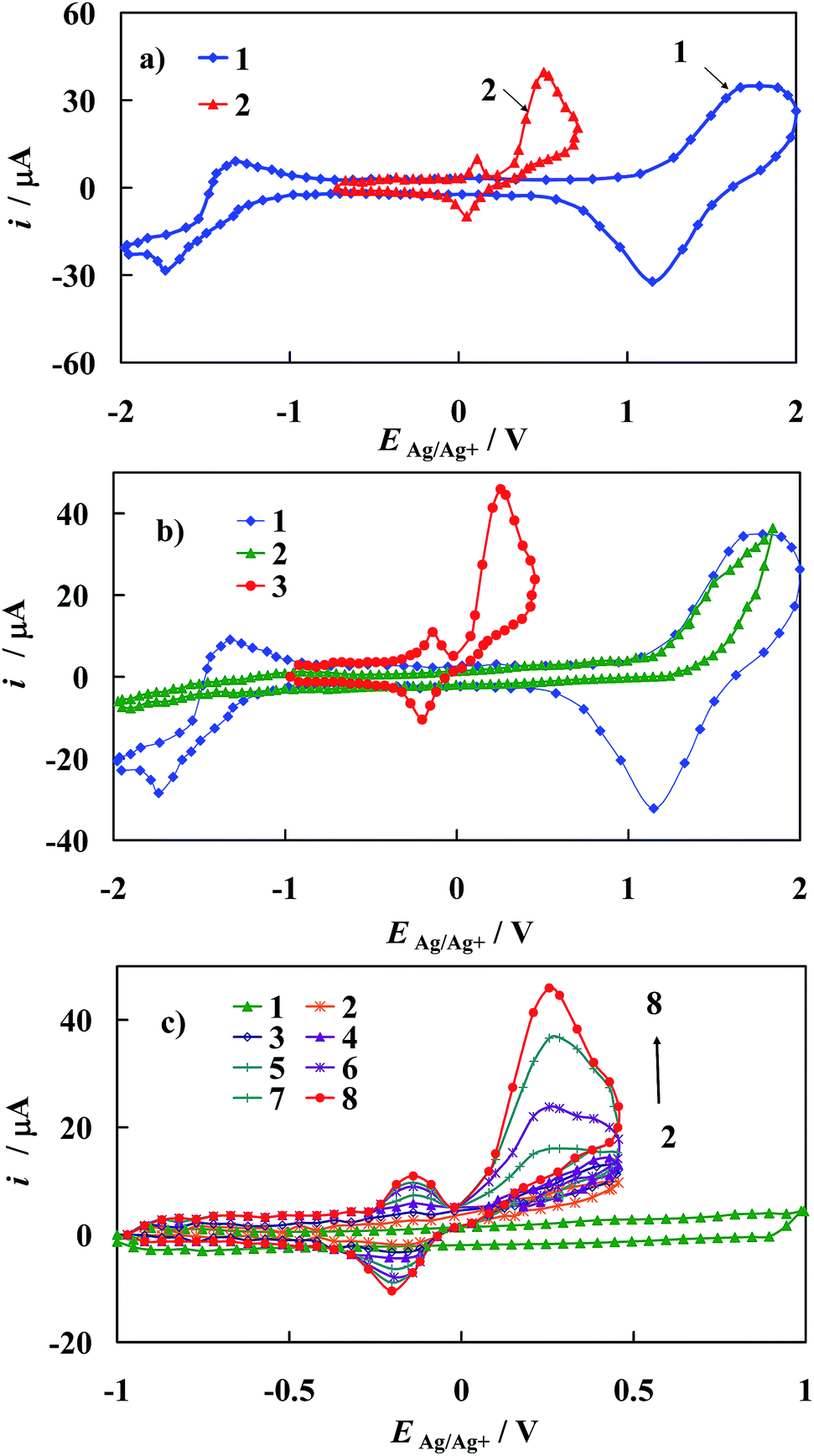 | ||
| Fig. 10 (a) Cyclic voltammograms of polycrystalline gold electrodes recorded in 0.1 M TBAClO4 in ACN (1) and with addition of (2) 500 μM of EP. (b) (1) Cyclic voltammograms of polycrystalline gold electrodes recorded in 0.1 M TBAClO4 in ACN and with addition of: (2) 1000 μM of the [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4; (3) the same as in (2) after addition of 500 μM EP. (c) Cyclic voltammograms of polycrystalline gold electrode recorded in 0.1 M TBAClO4 in ACN covered with 1000 μM of the Fe-complex (1) and (2) the same as in (1) after addition of 0.01 μM EP; (2) 0.1 μM EP, (3) 1 μM EP; (4) 10 μM EP; (5) 100 μM EP; (6) 200 μM EP; (7) 400 μM EP; (8) 500 μM EP. dE/dt = 0.1 V s−1. | ||
From comparison of the results presented in Fig. 10a and b, it is evident that a remarkable catalytic effect in epinephrine electrooxidation to o-epinephrinequinone can be achieved on gold electrode modified with [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4. This is manifested as a substantial shift of the corresponding voltammetric peaks towards the more negative direction accompanied by an increase in the current response at the potential of oxidation peak maximum in comparison with that of the bare gold electrode. Both these effects confirmed electrocatalytic improvement of the kinetics of the electrode reactions at electrochemically modified electrodes.52
The i–E curves recorded on the Au/1 modified electrode with increasing concentration of EP in the supporting electrolyte solution (Fig. 10c) suggest the presence of specific interactions of the epinephrine with the electrode material and/or with SAM of 1, which results in a decrease of the energy barrier for the oxidative electron transfer. This conclusion is supported by the Tafel slopes comprised between 140 mV for Au/1 electrode and 220 mV for the bare Au electrode.52
As mentioned above and visualized in Fig. 11a, the detection of epinephrine in the presence of other biogenic compounds like ascorbic acid on the bare gold electrode is precluded. For example, values of the peak separation (ΔEEP–AA) for the 500 μM EP concentration in the mixture with 5000 μM AA observed on unmodified gold electrode is only 0.05 V. As a result, the cyclic voltammetric responses of these two compounds practically overlap. Remarkably, this problem is removed when the modified gold electrode developed in this work is applied in analysis of the mixture containing EP and AA (Fig. 11b). In the latter case, the peak potential separation ΔEEP–AA increases to 0.45 V. Based on the crystallographic structure of [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4, it seems reasonable to conclude that its chemisorption on Au template leads to a SAM matrix with an excess of negative charge in the form of free electron pairs localized on N atoms of the Fe-complex in the chemisorbed layer. Thus, the electrode surface covered by the prepared [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex film should repel anionic species through electrostatic repulsion while cationic species should be electrostatically attracted. Taking into account the values of pKa = 9.9 and pKa = 4.1 of epinephrine and ascorbic acid, respectively,53 it is reasonable to conclude that under presented experimental conditions, EP exists in its cationic form while AA in its anionic form. It should be noted that the above given pKa values were determined in water, alas the absolute pKa values will be significantly higher in non-aqueous solvent,54,55 yet it will not affect final electrostatic forms of EP and AA. Thus, the positive potential shift of the EP peak current may be attributed to the electrostatic attraction between the positively charged EP and the negatively charged self-assembled layer. Moreover a repulsive interaction between the negatively charged of AA and the negatively charged SAM would be the reason for the movement of the peak current of AA towards the negative potential values, as is observed on applied modified electrode (Fig. 11b).
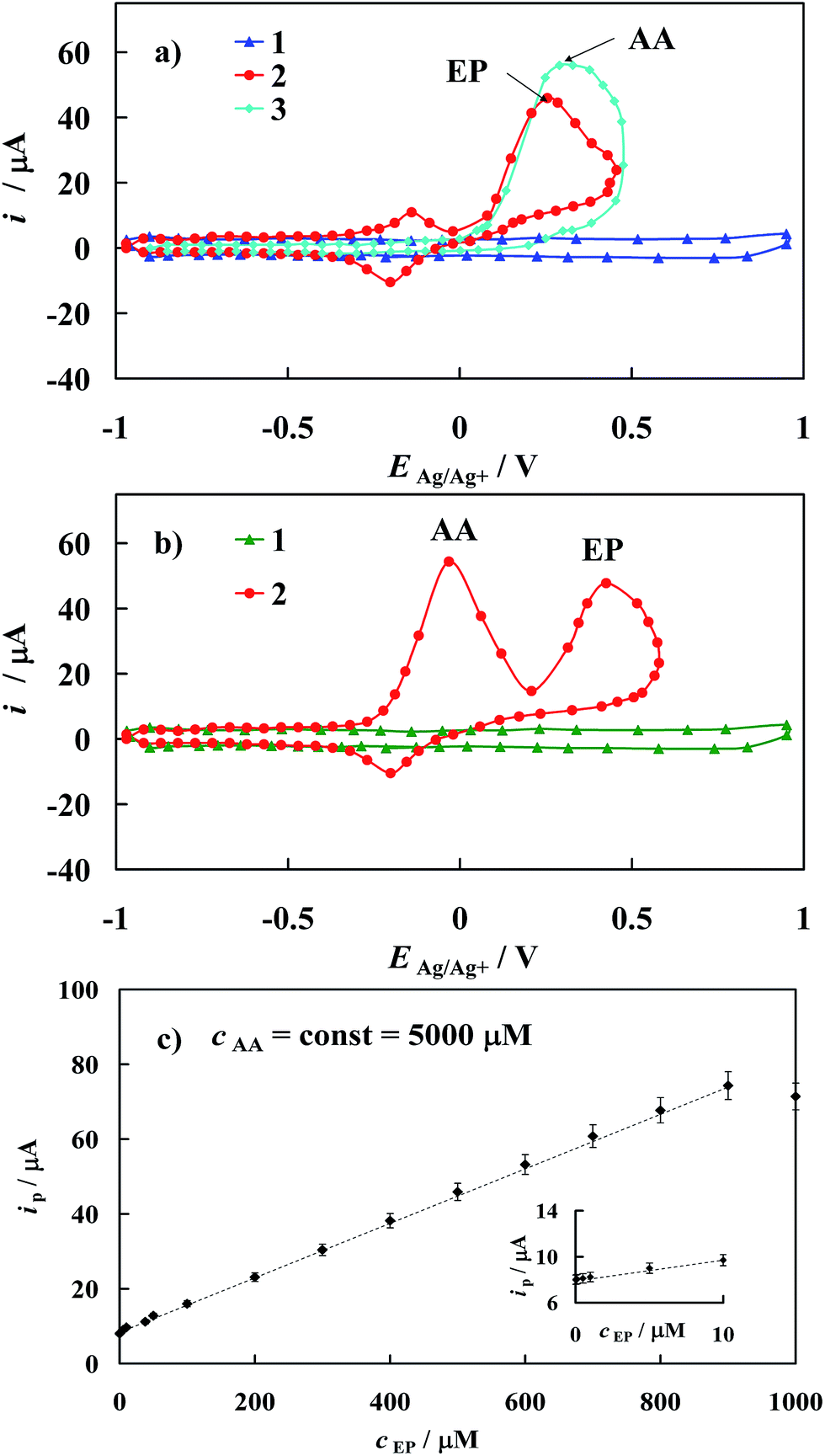 | ||
| Fig. 11 (a) Cyclic voltammograms recorded in 0.1 M TBAClO4 in ACN in the region of the electric double layer of polycrystalline gold electrode (1); and the same as in (1) after addition of 500 μM EP (2) and 5000 μM AA (3). dE/dt = 0.1 V s−1. (b) Cyclic voltammograms recorded in 0.1 M TBAClO4 in ACN in the region of the electric double layer of polycrystalline gold electrode covered with 1000 μM of the [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 (1) and the same as in (1) after addition of the mixture of 500 μM EP + 5000 μM AA. dE/dt = 0.1 V s−1. (c) The relationship of the peak current of epinephrine oxidation to epinephrinequinone in the presence of ascorbic acid at constant concentration (5000 μM) versus epinephrine concentration in 0.1 M TBAClO4 in ACN for modified gold electrodes with SAMs of Fe-complex; dE/dt = 0.1 V s−1, (R2 ≥ 0.9997). | ||
The latter behavior confirms that the modified gold electrode prepared in this paper may be successfully used for in vitro quantitative determination of EP in the presence of excess of AA. It should be emphasized as well that electrostatic interactions are reciprocal to dielectric constant of the solvent medium, therefore effect of solvent would be diminished, thus resulting in enhancement of interactions between adsorbed SAMs of [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 and EP/AA compounds. This may in turn contribute to the observed electrocatalytic oxidation effect.
Information on the detection sensitivity of new voltammetric sensor towards determination of EP in the presence of interfering AA was obtained from experiments performed by changing the concentration of EP with that of AA kept constant, Fig. 11c. Analysis was carried out over the range of 0.01 μM to 1000 μM of EP at constant AA concentration equal to 5000 μM. A linear relationship of the oxidation current versus epinephrine concentration (cEP) with the correlation coefficient R2 ≥ 0.9997 has been observed over the range of 0.01 μM to 900 μM, Fig. 11c and may be described by the following regression equations: ip [μM] = (845 ± 9) × 10−2 + (740 ± 8) × 10−4cEP [μM]. The detection sensitivity (ip/cEP) of the prepared sensor for epinephrine determination was determined as 2.83 A cm−2 M−1. Moreover, the detection limit (it should be emphasized that the lower value of the detection limit means the possibility of the detection of the lower analyte concentration) of epinephrine was estimated as 0.0074 μM according to the formula recommended by IUPAC: cL = k × sb/S (ref. 56) (where sb is the standard deviation of the current density recorded in the supporting electrolyte solution evaluated on the basis of 10 times repeated measurements, S is the slope of the linear ipvs. cEP dependence and k = 3 is a numerical constant).
One might also consider the effect of other interfering substances in the biological media. In the previous work36 we showed that both uric and ascorbic acids are effectively separated from dopamine when measured with Mn/Au composite electrode. Herein, Fe/Au composite was experimentally proved to be capable of measuring the concentration of epinephrine in the presence of AA. The common feature of both composite systems is that their sensing-like properties are dependent on the electrostatic interactions, therefore the acidity of detected compounds determines the potential values at which they are detected. Per analogy, one might expect that pKa = 4.1 and pKa = 5.4 values of ascorbic and uric acids are amenable for the successive separation of the latter ones from EP herein as well. One might consider though if dopamine (DA) and epinephrine – which belong to the same family – could be effectively separated i.e. that the former one would not hinder the detection of the latter one. Oxidation of both compounds at the bare gold electrode happens at E = 0.27 V/SCE (DA) and E = 0.31 V/SCE (EP) respectively, alas the value of the peak separation (ΔEDA–EP) is marginal. However, pKa values of DA and EP are 8.74 (ref. 57) and 9.90 (ref. 58) respectively. This means that both molecules would be present in their cationic form and would be positively attracted to the (1)/Au composite electrode. The most important however is that over an order of magnitude difference in acidity between EP and DA is enough for their effective separation to occur, hence EP can be successfully sensed in the presence of either AA, UA or DA.
The results observed prove that the modified gold electrode enhances the sensitivity towards EP detection since at the bare gold the linearity between the current density and the EP concentration was obtained in a narrower concentration range (0.1 μM to 500 μM) with the detection limit cL = 0.13 μM.31 Moreover, it is worth noting that both the range of concentrations over which the EP content can be reliably estimated on the basis of the above regression equation and the EP detection limit is comparable or even higher than those reported in some literature for different modified electrodes (for comparison see ref. 59). It should be however noted that the data presented in Table 1 from ESI† of ref. 59 are estimated to EP behavior in water solution, but as determination is made in vitro condition the replacement of water by ACN will have no effect on such a comparison.
3.4 Electrode reproducibility and stability of the modified electrode
In the reproducibility tests, it was found that the relative standard deviations (RDS) of ten replicate voltammetric responses of 500 μM epinephrine solution on the novel Au/Fe-complex SAM modified gold electrode was less than 3% indicating excellent reproducibility of the prepared sensor. The stability of the [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 modified electrode was checked daily during two weeks. Before each subsequent measurement, the electrically active composite was only washed with ACN. In two weeks, the voltammetric responses for the 500 μM epinephrine solution decreased by about 5% of their initial response.4. Conclusions
The results of the study demonstrate that the newly synthesized [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex effectively forms SAMs on gold electrodes and exhibits valuable properties with respect to the electrochemical activity of epinephrine – a compound of biological importance. We have shown that previously published, shielded, μ-phenoxo bridged ‘closed’ architecture of [Mn2(H2L)2](ClO4)2 is not mandatory to form stable composites at gold electrode, as herein studied Fe-complex exhibits different conformation of H3L ligand and thus structure. This finds reflection in different adsorption mode at the gold surface, yet still retains its electrocatalytic properties and thus offers a new insight into electrochemical modification of gold. We have also isolated and characterized H3L ligand and could thus demonstrate that not only complex 1 may be formed via direct reaction but also that the electrochemically mediated deposition of the sole ligand does not occur. It is crucial to note that present study paves the way towards design of a broader array of Schiff base metallosupramolecular architectures as electrochemical sensors towards neurotransmitters. The modified electrode shows good selectivity, sensitivity, reproducibility and high stability. There is no doubt that the modified electrode prepared in this paper is suitable for precise determination of EP concentration without any interference of AA. The prepared voltammetric sensor may be used – upon extraction of the analite from the real sample – for in vitro determination of epinephrine, as it is not important if the EP in such analysis is prepared in water or in ACN. What is of significant notice is that utilization of our sensor does not cause ACN to be introduced into the human body. The preparation procedure of the modified electrode with the Fe-complex SAM is simple and has promising application for detection of EP in physiological environment. An explanation of the detailed mechanism of EP oxidation on the electrode modified with the [Fe2(H3L)2(MeOH)2(μ-OMe)2](ClO4)4 complex, of its structure and interaction between obtained SAM layer and EP will be the task of our future study. Furthermore, isolation of H3L ligand will allow us to synthesize new architectures that comprise other d-metal ions, when the template approach was not successful.Acknowledgements
This research was carried out as a part of the Polish Ministry of Science and Higher Education project (Grant No. 0111/DIA/2012/41) in the frame of “Diamond Grant” programme. AG: I declare that I am a scholarship holder: (i) funded by the Polish National Science Center (Grant No. 2016/20/T/ST5/00187) in the frame of ETIUDA programme; (ii) within the project “Scholarship support for Ph.D. students specializing in majors strategic for Wielkopolska's development”, Sub-measure 8.2.2 Human Capital Operational Programme, co-financed by European Union under the European Social Fund as well as (iii) of The Adam Mickiewicz University Foundation in Poznań, 2014/2015 year edition. The Ministry of Scientific Research, Poland is gratefully acknowledged by T. Łuczak and K. Szymkowiak for financial support of this work.Notes and references
- K. E. Barrett, S. M. Barman, S. Boitano and H. L. Brooks, Ganong's review of medical physiology, McGraw-Hill e-Book, 24th edn, 2012 Search PubMed.
- M. Mueller, Clinical pharmacology: current topics and case studies, Springer, Vienna, 2008 Search PubMed.
- M. D. Hawley, S. V. Tatawawadi, S. Piekarski and R. N. Adams, Electrochemical studies of the oxidation pathways of catecholamines, J. Am. Chem. Soc., 1967, 89, 447 CrossRef CAS PubMed.
- J. Chen and C. S. Cha, Detection of dopamine In the presence of a large excess of ascorbic acid by using the powder microelectrode technique, J. Electroanal. Chem., 1999, 463, 93 CrossRef CAS.
- W. Ren, H. Q. Luo and N. B. Li, Simultaneous voltammetric measurements of ascorbic acid, epinephrine and uric acid at a glassy carbon modified with caffeic acid, Biosens. Bioelectron., 2006, 21, 1086 CrossRef CAS PubMed.
- A. Salimi, C. E. Banks and R. G. Compton, Abrasive immobilization of carbon nanotubes on basal plane pyrrolytic graphite electrode: application to the detection of epinephrine, Analyst, 2004, 129, 225 RSC.
- G. P. Jin, X. Peng and Y. F. Ding, The electrochemical modification of clenbuterol for paraffin-impregnated graphite electrode, Biosens. Bioelectron., 2008, 24, 1031 CrossRef CAS PubMed.
- F. C. Moraes, L. C. Golinelli, L. H. Mascaro and S. A. S. Machado, Determination of epinephrine in urine using multi-walled carbon nanotube modified with cobaltophtalocyanide in paraffin composite electrode, Sens. Actuators, B, 2010, 148, 492 CrossRef CAS.
- Y. Zeng, J. Yang and K. Wu, Electrochemistry and determination of epinephrine using a mesoporous Al-incorporated SiO2 modified electrode, Electrochim. Acta, 2008, 53, 4615 CrossRef CAS.
- X. Jiang and X. Lin, Immobilization of DNA on carbon fiber microelectrodes by using overoxidized polypyrrole template for selective detection of dopamine and epinephrine in the presence of high concentrations of ascorbic and uric acid, Analyst, 2005, 130, 391 RSC.
- B. Jin and H. Zhang, Nano-gold modified glassy carbon electrode for selective determination of epinephrine in the presence of ascorbic acid, Anal. Lett., 2002, 35, 1907 CrossRef CAS.
- N. F. Atta, A. Galal and S. M. Azab, Electrochemical determination of neurotransmitters using gold nanoparticles on nafion/carbon paste modified electrode, J. Electrochem. Soc., 2012, 159, H765 CrossRef CAS.
- N. F. Atta, A. Galal, F. M. Abu-Attia and S. M. Azab, Simultaneous determination of paracetamol and neurotransmitters in biological fluids using a carbon paste sensor modified with gold nanoparticles, J. Mater. Chem., 2011, 21, 13015 RSC.
- J. Li and X. Q. Lin, Electrodeposition of gold nanoclusters on overoxidized polypyrrole film modified glassy carbon electrode and its application for the simultaneous determination of epinephrine and uric acid under coexistence of ascorbic acid, Anal. Chim. Acta, 2007, 596, 222 CrossRef CAS PubMed.
- Y. Zhang, W. Ren and S. Zhang, Simultaneous determination of epinephrine, dopamine, ascorbic acid and uric acid by poly dopamine-nanogold composites modified electrode, Int. J. Electrochem. Sci., 2013, 8, 6839 CAS.
- W. Ren, H. Q. Luo and N. B. Li, Simultaneous voltammetric measurements of ascorbic acid, epinephrine and uric acid at a glassy carbon modified with caffeic acid, Biosens. Bioelectron., 2006, 21, 1086–1092 CrossRef CAS PubMed.
- S. M. Chen and K. C. Lin, The electrocatalytic properties of biological molecules using polymerized luminal film-modified electrodes, J. Electroanal. Chem., 2002, 523, 93 CrossRef CAS.
- F. Ni, Y. Wang, D. Zhang, F. Gao and M. Li, Electrochemical oxidation of epinephrine and uric acid at a layered double hydroxide film modified glassy carbon electrode and its application, Electroanalysis, 2009, 22, 1130 CrossRef.
- P. Kalimuthu and S. A. John, Simultaneous determination of epinephrine, uric acid and xanthine in the presence of ascorbic acid using an ultrathin film of 5-amino-1,3,4-thiadiazole modified electrode, Anal. Chim. Acta, 2009, 647, 97 CrossRef CAS PubMed.
- G. P. Jin, Q. Z. Chen, Y. F. Ding and J. B. He, Electrochemistry behaviour of adrenalin, serotonin and ascorbic acid at novel poly rutin modified paraffin-impregnated graphite electrode, Electrochim. Acta, 2007, 52, 2535 CrossRef CAS.
- Z. Yang, G. Hu, X. Chen, J. Zhao and G. Zhao, The nano-Au self assembled glassy carbon electrode for selective determination of epinephrine in the presence of ascorbic acid, Colloids Surf., B, 2007, 54, 230 CrossRef CAS PubMed.
- J. A Ni, H. X. Ju, H. Y. Chen and D. Leech, Amperometric determination of epinephrine with an osmium complex and Nafion double-layer membrane modified electrode, Anal. Chim. Acta, 1999, 378, 151 CrossRef.
- S. M. Chen and K. T. Peng, The electrochemical properties of dopamine, epinephrine, norepinephrine and their electrocatalytic reactions on cobalt(II) hexacyanoferrate films, J. Electroanal. Chem., 2003, 547, 179 CrossRef CAS.
- S. M. Chen and C. J. Liao, Preparation and characterization of osmium hexacyanoferrate films and their electrocatalytic properties, Electrochim. Acta, 2004, 50, 115 CrossRef CAS.
- S. M. Chen, C. J. Liao and V. S. Vasantha, Preparation and electrocatalytic properties of osmium oxide/hexacyanoruthenate films modified electrodes for catecholamines and sulphur oxoanions, J. Electroanal. Chem., 2006, 589, 15 CrossRef CAS.
- A. Ulman, Formation and structure of self-assembled monolayers, Chem. Rev., 1996, 96, 1533 CrossRef CAS PubMed.
- Ž. Petrović, M. Metikoš-Huković and R. Babić, Potential-assisted assembly of 1-dodecanethiol on polycrystalline gold, J. Electroanal. Chem., 2008, 623, 54 CrossRef.
- S.-F. Wang, D. Du and Q.-C. Zou, Electrochemical behaviour of epinephrine at L-cysteine self-assembled monolayers modified gold electrode, Talanta, 2002, 57, 687 CrossRef CAS PubMed.
- H.-M. Zhang, X.-L. Zhou, R.-T. Hui, N.-Q. Li and D.-P. Liu, Studies of the electrochemical behavior of epinephrine at a homocysteine self-assembled electrode, Talanta, 2002, 56, 1081 CrossRef CAS PubMed.
- N. F. Atta, A. Galal and E. H. El-Ads, A novel sensor of cysteine self-assembled monolayers over gold nanoparticles for the selective determination of epinephrine in presence of sodium dodecyl sulphate, Analyst, 2012, 137, 2658 RSC.
- T. Łuczak, Comparison of electrochemical oxidation of epinephrine in the presence of interfering ascorbic and uric acids on gold electrodes modified with S-functionalized compounds and gold nanoparticles, Electrochim. Acta, 2009, 54, 5863 CrossRef.
- T. Łuczak, Gold electrode modified with self-assembled layers made of sulphur compounds and gold nanoparticles used for selective electrocatalytic oxidation of catecholamine in the presence of interfering ascorbic and uric acid, Int. J. Electrochem., 2011, 1 CrossRef.
- T. Łuczak, Epinephrine oxidation in the presence of interfering molecules on gold and gold electrodes modified with gold nanoparticles and thiodipropionic acid in aqueous solution. A comparative study, Electroanalysis, 2009, 21, 2557 CrossRef.
- A. Gorczyński, M. Kubicki, D. Pinkowicz, R. Pełka, V. Patroniak and R. Podgajny, The first example of erbium triple-stranded helicates displaying SMM behavior, Dalton Trans., 2015, 16833 RSC.
- A. Gorczyński, M. Zaranek, S. Witomska, A. Bocian, A. R. Stefankiewicz, M. Kubicki, V. Patroniak and P. Pawluć, The cobalt(II) complex of a new tridentate Schiff-base ligand as a catalyst for hydrosilylation of olefins, Catal. Commun., 2016, 78, 71 CrossRef.
- A. Gorczyński, D. Pakulski, M. Szymańska, M. Kubicki, K. Bułat, T. Łuczak and V. Patroniak, Electrochemical deposition of the new manganese(II) Schiff-base complex on a gold template and its application for dopamine sensing in the presence of interfering biogenic compounds, Talanta, 2016, 149, 347 CrossRef PubMed.
- M. Sajid, M. K. Nazal, M. Mansha, A. Alsharaa, S. M. S. Jillani and C. Basheer, Chemically modified electrodes for electrochemical detection of dopamine in the presence of uric acid and ascorbic acid: a review, Trends Anal. Chem., 2016, 76, 15 CrossRef CAS.
- Agilent Technologies, CrysAlis PRO (Version 1.171.33.36d), Agilent Technologies Ltd, 2011 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Gualardi, Completion and refinement of crystal structures with SIR92, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- T. Łuczak, A comparative studies of adsorption of aliphatic amines at a polycrystalline gold electrode, Collect. Czech. Chem. Commun., 2005, 70, 2027 CrossRef.
- J. Nitschke, Construction, substitution and sorting of metallo-organic structures via subcomponent self-assembly, Acc. Chem. Res., 2007, 40, 103 CrossRef CAS PubMed.
- D. W. Mulder, E. M. Shepard, J. E. Meuser, N. Joshi, P. W. King, M. C. Posewitz, J. B. Broderick and J. W. Peters, Insights into [FeFe]-hydrogenase structure, mechanism and maturation, Structure, 2011, 19, 1038 CrossRef CAS PubMed.
- J. J. Griese, V. Srinivas and M. Högbom, Assembly of nonheme Mn/Fe active sites in heterodinuclear metalloproteins, JBIC, J. Biol. Inorg. Chem., 2014, 19, 759 CrossRef CAS PubMed.
- H. Houjou, M. Kanesato, K. Hiratani and D. Mandon, Structural and electrochemical studies of novel bis(m-methoxo)diiron complexes: observation of FeIIIFeIV and FeIVFeIV states resonating with phenoxyl radicals, Chem.–Eur. J., 2004, 10, 4576 CrossRef CAS PubMed.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Shape Maps and Polyhedral Interconversion Paths in Transition Metal Chemistry, Coord. Chem. Rev., 2005, 249, 1693 CrossRef CAS.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, Shape v. 2.0, Universitat de Barcelona, Barcelona, 2010 Search PubMed.
- A. Bielański, Inorganic Chemistry, PWN, Warsaw, 1999 Search PubMed.
- J. McMurry, Organic Chemistry, National Scientific Publishers, Warsaw, 2005 Search PubMed.
- G. Dodero, L. De Michieli, O. Cavalleri, R. Rolandi, L. Olivieri, A. Dacca and R. Parodi, L-Cysteine chemisorption on gold: an XPS and STM study, Colloids Surf., A, 2000, 175, 121 CrossRef CAS.
- A. J. Bard and L. K. Faulkner, Electrochemical methods: Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
- T. Łuczak, Electrocatalytic application of an overoxidized dopamine film prepared on a gold electrode surface for selective epinephrine sensing, Electroanalysis, 2008, 20, 1317 CrossRef.
- I. Kaljurand, A. Kutt, L. Soovali, T. Rodima, V. Maemets, I. Leito and I. A. Koppel, Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pKa units: unification of different basicity scales, J. Org. Chem., 2005, 70, 1019 CrossRef CAS PubMed.
- A. Kutt, I. Leito, I. Kaljurand, L. Soovali, V. M. Vlasov, L. M. Yagupolskii and I. A. Koppel, A comprehensive self-consistent spectrophotometric acidity scale of neutral Brønsted acids in acetonitrile, J. Org. Chem., 2006, 71, 2829 CrossRef PubMed.
- Analytical Methods Committee, Recommendation for definition, estimation and use of the detection limit, Analyst, 1987, 112, 199 RSC.
- B. J. F. Hill and D. C. Dyer, Pharmacological characterization of α-adrenoceptors in the bovine median caudal artery, Eur. J. Pharmacol., 1997, 339, 147 CrossRef CAS PubMed.
- E. Dempsey, A. Kennedy, N. Fay and T. McCormac, Investigations into heteropolyanions as electrocatalysts for the oxidation of adrenaline, Electroanalysis, 2003, 15, 1835 CrossRef CAS.
- T. Łuczak, M. Bełtowska-Brzezinska and R. Holze, Electrocatalytic activity of gold modified with gold nanoparticles and self-assembled layers of meso-2,3-dimercaptosuccinic acid for oxidation of epinephrine in the presence of ascorbic and uric acid, Electrochim. Acta, 2014, 123, 135 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Schematic representation of observed chemisorption modes as well as SEM and TEM characterization. CCDC 1407322 and 1482453. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra22028b |
| This journal is © The Royal Society of Chemistry 2016 |