
Effect of solvent ratio and counter ions on the morphology of copper nanoparticles and their catalytic application in β-enaminone synthesis†
Aravind L. Gajengia,
Takehiko Sasaki b and
Bhalchandra M. Bhanage*a
b and
Bhalchandra M. Bhanage*a
aDepartment of Chemistry, Institute of Chemical Technology, Matunga, Mumbai-400 019, India. E-mail: bm.bhanage@gmail.com; bm.bhanage@ictmumbai.edu.in; Fax: +91 33611020; Tel: +91 22 33612603
bDepartment of Complexity Science and Engineering, Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5, Kashiwanoha, Kashiwa, Chiba 277-8561, Japan
First published on 17th October 2016
Abstract
This work reports the synthesis of shape selective copper nanoparticles (NPs) using a microwave irradiation method, using diverse ratios of an ethylene glycol (EG)/water system. The solvent ratio of EG/water plays a pivotal role in the selective formation of CuO NPs. The alteration of the counter ion of the copper precursor leading to the selective synthesis of copper (CuO, Cu2O, Cu(OH)2) NPs has been observed. The synthesized copper NPs are well characterized using FEG-SEM, TEM, XRD, XPS, NH3-TPD and BET surface area. The prepared CuO NPs show high activity towards the synthesis of β-enaminones and β-enamino esters from 1,3 diketones and amines.
Introduction
Nanomaterials and microstructures have gained valuable importance, due to their distinctive structures and diverse properties, for optical, electrical, magnetic, thermal, catalytic and other extensive applications.1 Consequently, due to a wide variety of structures and properties, copper nanoparticles are considered for interesting applications in current research. Nowadays, copper NPs have found various applications in different fields such as catalysis,2 sensing,3 photocatalysis,4 fuel cells,5 lithium batteries,6 and water-splitting systems.7 There are different methods for the synthesis of copper NPs, such as the wet chemical method,8 biological methods,2a microwave methods,9 physical vapour deposition in ionic liquids,10 the solvothermal route,11 and the polyol method.12Microwave-assisted synthesis has attracted much attention because it has the advantages of being fast, simple, easily operated and more energy efficient.9a,13 Microwave synthesis enhances the kinetics of reactions by one or two orders of magnitude over conventional methods due to rapid initial heating and the generation of a localized high pressure zone at the reaction sites. The microwave process is more efficient than conventional processes for the preparation of various metal oxide NPs due to the shape selective synthesis of NPs.14,15 It is well known that the morphology of NPs varies as a result of a variation in the reaction parameters, such as the heating power of the microwave, the volume ratios of the mixed solvent and the counter ions.16,17 A mixed solvent system of EG and water has been used for the morphology controlled synthesis of CuO superstructures.18 There are some methods where CuO was synthesized using a mixed solvent of EG and water.3,18 Recently, Zhou et al. synthesized Cu2O/CuO microparticles, using EG/water as a mixed solvent in the autoclave, using copper acetate as a precursor.3 In this protocol they used only one volume ratio of EG/water, i.e., 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and observed the formation of Cu2O/CuO microparticles.
:1, and observed the formation of Cu2O/CuO microparticles.
β-Enaminones and β-enamino esters are important starting materials for the synthesis of organic compounds.19 These compounds are used for synthesis of heterocycles,20 nitrogen containing compounds,21 pharmaceutical drugs as anticonvulsants,22 natural alkaloids,23 and antibacterial compounds.24 Various methods have been developed for the synthesis of enaminones, such as the condensation of carbonyl compounds with amines using various catalysts, such as CoCl2,25 Yb(OTf)3,26 Cu NPs,27 Ag NPs,28 perchlorates,29 InBr3,30 and tungstophosphoric acid,31 and ultrasonic methods.32 These reported methods have one or more disadvantages, such as the use of moisture sensitive metal triflates and difficulties in the work up,26 the use of harmful reagents,29 or the use of homogenous catalysts.25 There is a need to develop an efficient catalytic method for the synthesis of β-enaminones, which operates under mild reaction conditions.
In this report, we synthesized and characterized CuO NPs under microwave irradiation, using different volume ratios of EG/water as solvent. The effect of various copper precursors leads to the shape selective formation of copper NPs, such as CuO, Cu2O, and Cu(OH)2. These synthesized CuO NPs are acidic in nature and show excellent catalytic activity towards the synthesis of β-enaminones and β-enamino esters from 1,3 di-ketones and amines under mild conditions.
Experimental
Chemicals and reagents
Copper acetate [Cu(CH3COO)2·H2O], copper chloride [CuCl2·2H2O], copper nitrate [Cu(NO3)2] and ethylene glycol were procured from S.D. Fine Chemicals Pvt. Ltd. India and were used as received without further purification.Preparation of CuO NPs
For the synthesis of CuO NPs, 200 mg of copper acetate was dissolved in different volume ratios of EG/water in mL (i.e., 1:9, 3:7, 5:5, 7:3, and 9:1), transferred to a 100 mL glass beaker and kept inside a microwave oven for 4 min at 600 W, with the on–off mode of the microwave oven having a time interval of 30 s. After microwave heating, the colour of the reaction mixture changes to a blue to a black precipitate, indicating the formation of CuO NPs. The product was separated through centrifugation at 7000 rpm for 10 min, and washed with distilled water and ethanol several times. The obtained product was dried in an oven and used for further characterization. The CuO NPs prepared at different volume ratios of EG/water, i.e., 1:9, 3:7, 5:5, 7:3, 9:1 in mL, are labelled as CuO (a), CuO (b), CuO (c), CuO (d) and CuO (e) respectively.
Preparation of Cu2O NPs
For the synthesis of Cu2O NPs, 188 mg of Cu(NO3)2 was dissolved in 3:7 and 7:3 volume ratios of EG/water in mL, transferred to a 100 mL glass beaker and kept inside a microwave oven for 4 min at 600 W with the on–off mode of the microwave oven having a time interval of 30 s. After microwave heating, the colour of the reaction mixture changes from a blue to a red precipitate, indicating the formation of Cu2O NPs. The product was separated through centrifugation at 7000 rpm for 10 min and washed with distilled water and ethanol several times. The obtained product was dried in an oven and used for further characterization.
Preparation of Cu(OH)2 NPs
For the synthesis of Cu(OH)2 NPs, 170 mg of copper chloride was dissolved in 3:7 and 7:3 volume ratios of EG/water in mL, transferred to a 100 mL glass beaker and kept inside a microwave oven for 4 min at 600 W with the on–off mode of the microwave oven having a time interval of 30 s. After microwave heating, the colour of the reaction mixture changes from a blue to a green precipitate, indicating the formation of Cu(OH)2 NPs. The product was separated through centrifugation at 7000 rpm for 10 min and washed with distilled water and ethanol several times. The obtained product was dried in an oven and used for further characterization.
Characterization methods for CuO, Cu2O, and Cu(OH)2 NPs
The prepared CuO, Cu2O and Cu(OH)2 NPs were characterized using various analytical techniques. An X-ray diffractometer (Shimadzu XRD-6100, using Cu Kα = 1.54 Å) with a scanning rate of 2° per min and a 2 theta (θ) angle ranging from 20° to 80° with a current of 30 mA and a voltage of 40 kV was used, and field emission gun-scanning electron microscopy (FEG-SEM) analysis was done using a Tescan MIRA 3 model with a secondary electron (SE) detector between 10.0 kV and 20.0 kV. For transmission electron microscopy (TEM), a Philips CM 200 was used at an operating voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) was carried out using a PHI 5000 Versa Probe Scanning ESCA Microprobe, and the Brunauer–Emmett–Teller (BET) surface area was analyzed using an ASAP 2010 (Micromeritics, USA) instrument. NH3-temperature programmed desorption (NH3-TPD) was recorded using a Thermo Scientific TPDRO 1100 machine.The general procedure for the synthesis of β-enaminones and β-enamino esters
The experiments were carried out in a 25 mL reaction vial. In a typical reaction procedure, the CuO NPs, as catalyst, were introduced into a reaction vial containing acetylacetone (1 mmol) and an amine (1 mmol). The reaction mixture was stirred at a temperature of 60 °C for 9 h. The progress of the reaction was monitored using TLC. After completion of the reaction, the catalyst was recovered using filtration. The mixture was concentrated under vacuum. The residue was purified using chromatography to afford the desired compound. All products are well known in the literature and were characterized using GC-MS (Shimadzu QP 2010), and were also compared with authentic samples.Results and discussion
Effect on the morphology of CuO NPs of different ratios of EG/water
Initially, by using copper acetate as the metal precursor, we started an investigation into the synthesis of CuO NPs with different solvent volume ratios of EG/water under microwave irradiation at 600 W for 4 minutes. A variation in the volume ratio of EG/water leads to a change in morphology (Fig. 1). Using a 1:9 ratio of EG/water, the aggregation of sphere like CuO NPs was observed in SEM images (Fig. 1a). When the composition ratio of EG/water changes to 3:7, spindle shaped CuO NPs are observed (Fig. 1b). A disturbed spindle shaped morphology was observed when a 5:5 ratio of EG/water was used (Fig. 1c). Interestingly, a flower-like morphology was observed in the SEM as well as the TEM images when a 7:3 ratio of EG/water was used (Fig. 1d and f). In the case of a 9:1 ratio of EG/water, spherical CuO microparticles are observed (Fig. 1e). From this, it was concluded that the volume ratio of EG/water controls the shape and size of the CuO NPs.
 | ||
| Fig. 1 FEG-SEM images of CuO NPs synthesized at different volume ratios of EG/water in mL: (a) 1:9, (b) 3:7, (c) 5:5, (d) 7:3, and (e) 9:1. (f) A TEM image of CuO NPs from a 7:3 ratio of EG/water, synthesized at 600 W for 4 min. | ||
Following the successful synthesis of the NPs, we further characterized these NPs using various techniques such as XRD, XPS and NH3-TPD. The XRD pattern of the CuO NPs synthesized at volume ratios of 3:7 and 7:3 in mL of EG/water (Fig. 2) shows the lattice planes of (−111) and (200) planes, with corresponding Bragg angles of 35.6° and 38.8°, which matches the characteristic peaks of pure monoclinic CuO crystallites (JCPDS card 05-0661).33 The XRD patterns of CuO NPs synthesized at different volume ratios of EG/water also match the standard (ESI-S1,† Fig. 1).
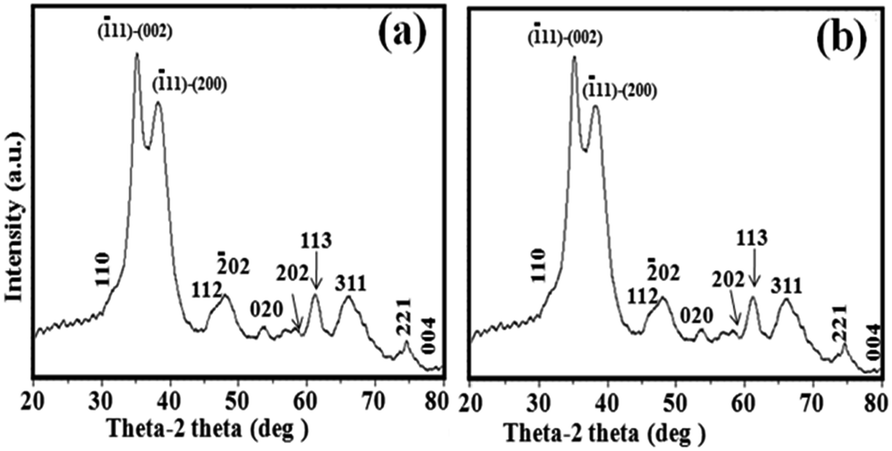 | ||
| Fig. 2 XRD patterns of CuO NPs with a (a) 3:7 and (b) 7:3 ratio of EG/water as the solvent system. | ||
For the surface analysis of the synthesised NPs, we further characterized them using XPS analysis. The presence of Cu and O elements was confirmed through XPS analysis (Fig. 3a). The peak observed at a binding energy of 529.5 eV corresponds to O1s and confirms O2− from CuO (Fig. 3b). The peaks observed at binding energies of 933.6 eV and 952.9 eV correspond to Cu2p3/2 and Cu2p1/2 (Fig. 3c), which can be attributed to CuO and are well reported in the literature.34 Along with these, Cu2p3/2 and Cu2p1/2 satellite peaks are also observed at ∼940 eV and ∼959.8 eV, which are characteristic of partially filled d-orbitals (3d9 in this case from Cu2+).35
 | ||
| Fig. 3 XPS spectra of CuO NPs: (a) survey spectrum, (b) O1s region and (c) Cu2p region synthesized at 7:3 of EG/water. | ||
Effect of counter ion in the synthesis of CuO, Cu2O and Cu(OH)2
Further studies on the effects of morphology were conducted by changing the counter ions of the Cu metals at 3:7 and 7:3 ratios of EG/water under microwave irradiation. Surprisingly, it was observed that the use of copper nitrate as a precursor resulted in the formation of Cu2O NPs (Fig. 4a and b). The use of copper chloride as a precursor affords the formation of disturbed spherical Cu(OH)2 particles when a 3:7 ratio of EG/water was used (Fig. 4c) and a fibrous, flake-like morphology was obtained when a 7:3 ratio of EG/water was used (Fig. 4d).
 | ||
| Fig. 4 SEM images of Cu NPs when changing the metal precursor: (a) and (b) Cu2O NPs from Cu(NO3)2, and (c) and (d) Cu(OH)2 microparticles synthesised from CuCl2. | ||
The XRD patterns of the Cu2O and Cu(OH)2 NPs also match with the reported JCPDS data (ESI S1,† Fig. 2).36,37 Hence, the ratio of EG/water and the precursor control the morphology of the particles in the selective formation of copper NPs. The NH3-TPD data from the synthesized CuO NPs was recorded and it was observed that the acidity of the NPs varies with the morphology (Fig. 5). The amount of NH3 desorbed for CuO (a), CuO (b), CuO (c), CuO (d) and CuO (e) was 1817, 2502, 2751, 9813 and 2106 μmol g−1, respectively. The CuO NPs (d), having flower-like morphology, showed the highest NH3 desorption (9813 μmol g−1), hence they are expected to have the highest acidity.
 | ||
| Fig. 5 Comparative NH3-TPD data for the synthesized CuO NPs. | ||
The BET-specific surface area of a catalyst is an important parameter for high catalytic activity, and the observed BET specific surface areas for CuO (a), CuO (b), CuO (c), CuO (d) and CuO (e) are 98, 115, 118, 123 and 25 m2 g−1, respectively.
Catalytic application to β-enaminones and β-enamino esters of CuO NPs
To investigate the catalytic application of the synthesized NPs, the reaction of the 1,3-dicarbonyl compound 1a and amine 2b was carried out to synthesize the β-enaminone 3aa. Initially, the reaction was carried out without catalyst and it was observed that only trace amounts of the product 3aa were obtained (Table 1, entry 1). Then, we screened various CuO NPs (Table 1, entries 2–6), and among them the CuO NPs (d) show the highest catalytic activity and provide a yield of up to 58% (Table 1, entry 5). The higher catalytic activity of the flower-shaped CuO (d) may be due to the higher acidity and BET specific surface area (123 m2 g−1) compared to the other catalysts. The sphere-like CuO (a) and micro-sized CuO (e) show a lower acidity and BET specific surface area and, due to this, give a lesser yield of the product 3aa (Table 1, entries 2 and 6). Also, the spindle shaped CuO (b) gave a lesser yield of product 3aa due to a lower acidity and BET surface area when compared to CuO (d) (Table 1, entry 3). The disturbed spindle shaped CuO (c) shows a higher acidity and BET specific surface area than CuO (b) and shows a higher yield of product 3aa than that of catalyst CuO (b) (Table 1, entry 4). So the acidity and specific surface area are important for the catalytic activity of CuO NPs.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Solvent | Temp. (°C) | Time (h) | Yieldb [%] |
| a Reaction conditions: acetylacetone (1.0 mmol), aniline (1.0 mmol), and CuO NPs (mol% with respect to the starting material), under a N2 atmosphere.b GC yield. The GC yield was quantified with an external standard, using methyl 4-(phenylamino)pent-3-en-2-one. | ||||||
| Effect of catalyst screening | ||||||
| 1 | — | Methanol | 60 | 9 | Trace | |
| 2 | (a) | 5 | Methanol | 60 | 9 | 38 |
| 3 | (b) | 5 | Methanol | 60 | 9 | 45 |
| 4 | (c) | 5 | Methanol | 60 | 9 | 50 |
| 5 | (d) | 5 | Methanol | 60 | 9 | 58 |
| 6 | (e) | 5 | Methanol | 60 | 9 | 39 |
|
||||||
| Effect of catalyst loading | ||||||
| 7 | (d) | 1.5 | Methanol | 60 | 9 | 50 |
| 8 | (d) | 2.5 | Methanol | 60 | 9 | 64 |
| 9 | (d) | 3.0 | Methanol | 60 | 9 | 60 |
| 10 | (d) | 7.5 | Methanol | 60 | 9 | 57 |
|
||||||
| Effect of time | ||||||
| 11 | (d) | 2.5 | Methanol | 60 | 3 | 51 |
| 12 | (d) | 2.5 | Methanol | 60 | 6 | 57 |
| 13 | (d) | 2.5 | Methanol | 60 | 12 | 60 |
|
||||||
| Effect of solvent | ||||||
| 14 | (d) | 2.5 | Ethanol | 60 | 9 | 68 |
| 15 | (d) | 2.5 | Acetonitrile | 60 | 9 | 33 |
| 16 | (d) | 2.5 | Toluene | 60 | 9 | 39 |
| 17 | (d) | 2.5 | — | 60 | 9 | 83 |
|
||||||
| Effect of temperature | ||||||
| 18 | (d) | 2.5 | — | RT | 9 | 44 |
| 19 | (d) | 2.5 | — | 40 | 9 | 63 |
| 20 | (d) | 2.5 | — | 50 | 9 | 70 |
| 21 | (d) | 2.5 | — | 60 | 9 | 83 |
| 22 | (d) | 2.5 | — | 70 | 9 | 84 |

In order to further optimize the reaction conditions, the CuO NPs (d) were chosen for the synthesis of β-enaminones and β-enamino esters, due to their high activity. In a study of catalyst loading, an increase in catalyst from 1.5 to 2.5 mol% increases the yield of 3aa (Table 1, entries 7 and 8). However, an increase from 3.0 to 7.5 mol% slightly decreases the yield of product 3aa (Table 1, entries 8–10). Importantly, to check the optimum time for the reaction, this reaction was carried out for 3 h, 6 h, 9 h and 12 h, and it was observed that with an increase in time from 3 h to 9 h, there was an increase in the yield of product 3aa, and with a further increase in the time to 12 h, there was no effect on the yield of the product 3aa (Table 1, entries 11–13). Next, in the solvent study, the use of solvents such as methanol, ethanol, acetonitrile and toluene gave a lesser yield than a reaction carried out under neat conditions (Table 1, entries 14–17). Finally, temperature studies were done at RT, 40 °C, 50 °C, 60 °C and 70 °C. With an increase in the temperature from RT to 60 °C, there was an increase in the yield of product 3aa, and with a further increase in the temperature, there was a slight change in the yield of product 3aa (Table 1, entries 18–22). So, the final optimum reaction conditions were a catalyst amount of 2.5 mol% and a time of 9 h, at 60 °C under solvent free conditions.
On the basis of the above optimized reaction conditions, the scope of substrates for β-enaminone and β-enamino ester synthesis was evaluated (Table 2). The condensation of aniline 1a with acetylacetone 2a provided an excellent yield of 3aa (Table 2, entry 1). The effect of electron donating and withdrawing groups on aniline was studied. It was found that the electron donating group –Me and withdrawing group –F produced the corresponding products 3ab–3ac in good yield (Table 2, entries 2 and 3). Aliphatic primary amines, such as 2d, 2e and 2f, give the desired products 3ad–3af (Table 2, entries 4–6). Heterocyclic secondary amines such as 2g and 2h give an excellent yield of the products 3ag–3ah (Table 2, entries 7–8). Next, the reaction was carried out, with a change of the 1,3-carbonyl compound to ethylacetoacetate 1b and methylacetoacetate 1c, with various amines. The reaction of 1b with 2a, 2e, 2f, 2g and 2h gives a good to excellent yield of products 3ba–3bh (Table 2, entries 9–13). Finally, the reaction of 1c with 2a, 2f and 2h gives a corresponding yield of the products 3ca–3ch (Table 2, entries 14–16).
|
||||
|---|---|---|---|---|
| Entry | Dicarbonyl compound | Amine | Product | Yieldb [%] |
| a Reaction conditions: 1,3-dicarbonyl compound (1.0 mmol), amine (1.0 mmol), and CuO NPs (2.5 mol%), solvent free, at 60 °C for 9 h under a N2 atmosphere.b isolated yield. | ||||
| 1 | 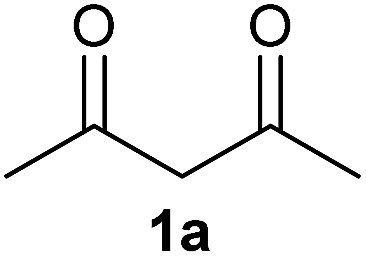 |
 |
 |
83 |
| 2 | 1a |  |
 |
80 |
| 3 | 1a |  |
 |
78 |
| 4 | 1a | 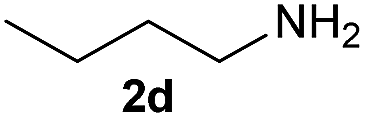 |
 |
93 |
| 5 | 1a | 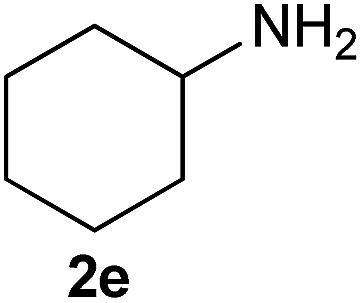 |
 |
97 |
| 6 | 1a |  |
 |
98 |
| 7 | 1a |  |
 |
90 |
| 8 | 1a |  |
 |
95 |
| 9 | 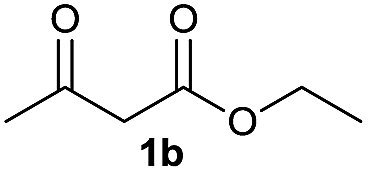 |
2a |  |
78 |
| 10 | 1b | 2e |  |
75 |
| 11 | 1b | 2f |  |
90 |
| 12 | 1b | 2g |  |
68 |
| 13 | 1b | 2h | 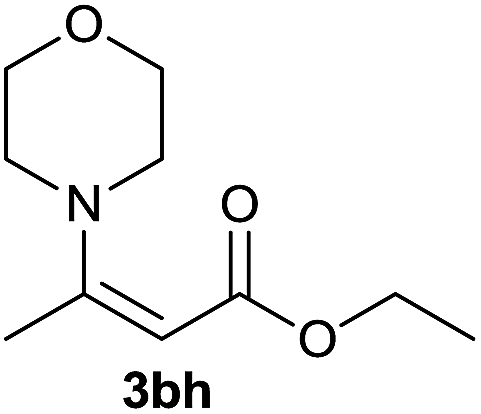 |
80 |
| 14 |  |
2a | 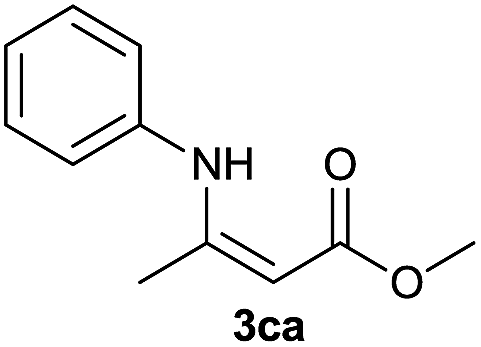 |
70 |
| 15 | 1c | 2f |  |
98 |
| 16 | 1c | 2h |  |
70 |

After a detailed substrate study, we moved towards a study into the recyclability of the heterogeneous catalytic system. The recyclability of the CuO NP catalyst was studied for the synthesis of the β-enaminone 3aa under optimised reaction conditions. After the completion of the reaction, the catalyst was separated through simple filtration techniques. The catalyst was washed with distilled water and ethanol, dried under vacuum and then reused for successive runs. Importantly, the catalyst was found to be effective for up to three consecutive runs without any significant loss in its catalytic activity (Fig. 6). After the recycling study, the catalyst was analysed using TEM (Fig. 7a) and it was observed that there is a slightly change in the morphology of the catalyst. In the XRD pattern, it was observed that there is not much change in the pattern (Fig. 7b). A comparison of the present catalytic system shows that it takes less time than some of the reported protocols, as shown in Table 3. A proposed mechanism for the formation of β-enaminones and β-enamino esters is shown in Scheme 1. In this, the carbonyl groups of the 1,3-dicarbonyl compound interact with CuO NPs to form the intermediate A which further interacts with the amine to give the intermediate B, which on the elimination of water gives the product β-enaminone.
 | ||
| Fig. 6 Recyclability in the synthesis of β-enaminones from 1,3 dicarbonyl compounds and amines catalysed using CuO NPs. Reaction conditions: 1,3-dicarbonyl compound (1 mmol), and amine (1 mmol), at 60 °C for 9 h, bGC yield. The GC yield was quantified with an external standard using methyl 4-(phenylamino)pent-3-en-2-one. | ||
 | ||
| Fig. 7 (a) TEM and (b) XRD analysis of the CuO NPs after recycling. | ||
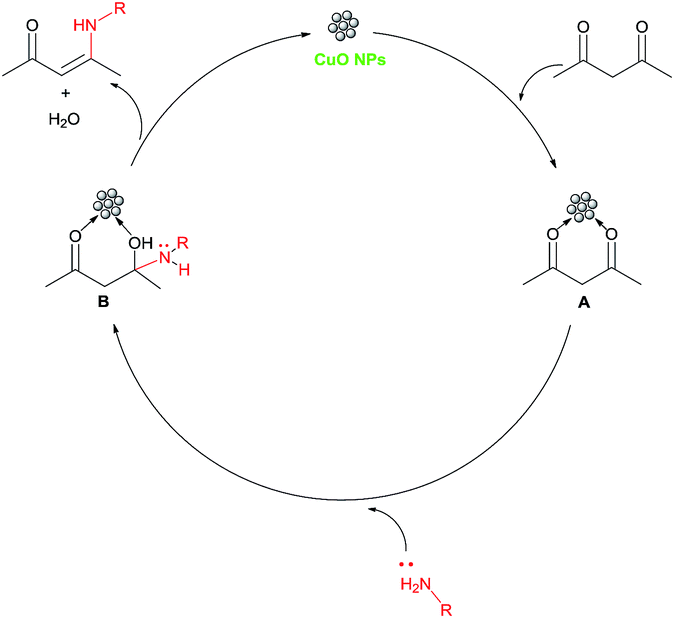 | ||
| Scheme 1 Reaction mechanism for the synthesis of β-enaminones catalysed using CuO NPs. | ||
Conclusions
In summary, we have prepared shape selective and catalytically active CuO NPs using different volume ratios of EG/water under a microwave irradiation method. The change in morphology of the CuO NPs with changes in the volume ratio of EG/water was observed. Interestingly, by changing the counter ion of copper, a change in the Cu NPs were observed; for example, Cu(OAc)2 provides CuO, Cu(NO3)2 provides Cu2O and CuCl2 provides Cu(OH)2. Additionally, the synthesized CuO shows high catalytic activation for the synthesis of β-enaminones and β-enamino esters. The developed methodology proceeds under solvent free conditions, utilizes mild reaction conditions and is recyclable for up to three cycles.Characterisation data for the products
4-(Phenylamino)pent-3-en-2-one (Table 2, entry 1)39
1H NMR (400 MHz, CDCl3) δ 12.45 (s, 1H), 7.31 (m, 2H), 7.17 (m, 1H), 7.10 (d, J = 7.6 Hz, 2H), 5.17 (s, 1H), 2.09 (s, 3H), 1.98 (s, 3H). GC-MS (EI, 70 eV): m/z (%) = 176 (6) [M+], 175 (45), 160 (100), 132 (56), 118 (38), 77 (35), 51 (19), 44 (39).4-(Cyclohexylamino)pent-3-en-2-one (Table 2, entry 5)39
1H NMR (400 MHz, CDCl3) δ 10.95 (s, 1H), 4.88 (s, 1H), 3.35 (s, 1H), 1.96 (s, 3H), 1.91 (s, 3H), 1.83 (s, 2H), 1.74 (s, 2H), 1.55 (d, J = 9.6 Hz, 1H), 1.36–1.21 (m, 5H). GC-MS (EI, 70 eV): m/z (%) = 182 (16) [M+], 181 (76), 166 (30), 138 (61), 100 (78), 84 (83), 58 (100), 43 (49).4-(Benzylamino)pent-3-en-2-one (Table 2, entry 6)40
1H NMR (400 MHz, CDCl3) δ 11.14 (s, 1H), 7.32 (d, J = 7.1 Hz, 2H), 7.25 (t, J = 7.5 Hz, 3H), 5.03 (s, 1H), 4.44 (d, J = 6.3 Hz, 2H), 2.02 (s, 3H), 1.90 (s, 3H). GC-MS (EI, 70 eV): m/z (%) = 190 (9) [M+], 189 (48), 174 (33), 146 (26), 105 (12), 91 (100), 65 (19), 42 (17).Ethyl 3-(cyclohexylamino)but-2-enoate (Table 2, entry 10)32
1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 4.37 (s, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.29 (d, J = 5.3 Hz, 1H), 1.91 (s, 3H), 1.75–1.53 (m, 3H), 1.31–1.19 (m, 10H). GC-MS (EI, 70 eV): m/z (%) = 212 (11) [M+], 211 (31), 182 (7), 166 (25), 130 (100), 122 (35), 84 (96), 55 (31), 41 (39).Acknowledgements
The author Aravind L. Gajengi is thankful to the University Grand Commission-Basic Research Programme (UGC-BSR) for providing a fellowship.References
- (a) C. H. Kuo and M. H. Huang, Nano Today, 2010, 5, 106–116 CrossRef CAS; (b) R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- (a) M. Nasrollahzadeh, S. M. Sajadi and A. R. Vartooni, J. Colloid Interface Sci., 2015, 459, 183–188 CrossRef CAS PubMed; (b) X. Zhang, G. Wang, M. Yang, Y. Luan, W. Dong, R. Dang, H. Gao and J. Yu, Catal. Sci. Technol., 2014, 9, 3082–3089 RSC.
- L. J. Zhou, Y. C. Zou, J. Zhao, P. P. Wang, L. L. Feng, L. W. Sun, D. Jun Wang and G. D. Li, Sens. Actuators, B, 2013, 188, 533–539 CrossRef CAS.
- A. Singhal, M. R. Pai, R. Rao, K. T. Pillai, I. Lieberwirth and A. K. Tyagi, Eur. J. Inorg. Chem., 2013, 2640–2651 CrossRef CAS.
- J. K. Lee, J. Choi, S. J Kang, J. M. Lee, Y. Tak and J. Lee, Electrochim. Acta, 2007, 52, 2272–2276 CrossRef CAS.
- S. Venkatachalam, H. Zhu, C. Masarapu, K. Hung, Z. Liu, K. Suenaga and B. Wei, ACS Nano, 2009, 3, 2177–2184 CrossRef CAS PubMed.
- D. Barreca, P. Fornasiero, A. Gasparotto, V. Gombac, C. Maccato, T. Montini and E. Tondello, ChemSusChem, 2009, 2, 230–233 CrossRef CAS PubMed.
- Y. Xu, D. Chen, X. Jiao and K. Xue, J. Phys. Chem. C, 2007, 111, 16284–16289 CAS.
- (a) M. A. Bhosale, K. D. Bhatte and B. M. Bhanage, Powder Technol., 2013, 235, 516–519 CrossRef CAS; (b) M. A. Bhosale and B. M. Bhanage, RSC Adv., 2014, 4, 15122–15130 RSC; (c) M. A. Bhosale, T. Sasaki and B. M. Bhanage, Catal. Sci. Technol., 2014, 12, 4274–4280 RSC.
- K. Richter, A. Birkner and A. V. Mudring, Angew. Chem., Int. Ed., 2010, 49, 2431–2435 CrossRef CAS PubMed.
- S. K. Li, C. H. Li, F. Z. Huang, Y. Wang, Y. H. Shen, A. J. Xie and Q. Wu, J. Nanopart. Res., 2011, 13, 2865–2874 CrossRef CAS.
- Z. C. Orel, A. Anzlovar, G. Drazic and M. Zyigon, Cryst. Growth Des., 2007, 2, 453–458 Search PubMed.
- T. Krishnakumar, R. Jayaprakash, M. Parthibavarman, A. R. Phani, V. N. Singh and B. R. Mehta, Mater. Lett., 2009, 63, 896–898 CrossRef CAS.
- Y. Mina, T. Wang and Y. C. Chena, Appl. Surf. Sci., 2010, 257, 132–137 CrossRef.
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chem.–Eur. J., 2005, 11, 440–452 CrossRef CAS PubMed.
- P. Zhu, J. Zhang, Z. Wu and Z. Zhang, Cryst. Growth Des., 2008, 9, 3148–3153 Search PubMed.
- R. Wu, Z. Ma, Z. Gu and Y. Yang, J. Alloys Compd., 2010, 504, 45–49 CrossRef CAS.
- S. Bhuvaneshwari and N. Gopalakrishnan, J. Colloid Interface Sci., 2016, 480, 76–84 CrossRef CAS PubMed.
- E. Rafiee, M. Joshaghani, S. Eavani and S. Rashidzadeh, Green Chem., 2008, 10, 982–989 RSC.
- A. Noole, M. Borissova, M. Lopp and T. Kanger, J. Org. Chem., 2011, 76, 1538–1545 CrossRef CAS PubMed.
- C. Alan, A. C. Spivey, R. Srikaran, C. M. Diaper, J. David and D. Turner, Org. Biomol. Chem., 2003, 1, 1638–1640 Search PubMed.
- I. O. Edafiogho, K. V. Ananthalakshmi and S. B. Kombian, Bioorg. Med. Chem., 2006, 14, 5266–5272 CrossRef CAS PubMed.
- G. Li, K. Watson, R. W. Buckheit and Y. Zhang, Org. Lett., 2007, 9, 2043–2046 CrossRef CAS PubMed.
- T. Mahmud, R. Rehman, A. Gulzar, A. Khalid, J. Anwar, U. Shafique, W. Zaman and M. Salman, Arabian J. Chem., 2010, 3, 219–224 CrossRef CAS.
- Z. H. Zhang and J. Y. Hu, J. Braz. Chem. Soc., 2006, 17, 1447–1451 CrossRef CAS.
- F. Epifano, S. Genovese and M. Curinib, Tetrahedron Lett., 2007, 48, 2717–2720 CrossRef CAS.
- M. Kidwai, S. Bhardwaj, N. K. Mishra, V. Bansal, A. Kumar and S. Mozumdar, Catal. Commun., 2009, 10, 1514–1517 CrossRef CAS.
- K. D. Bhatte, P. J. Tambade, K. P. Dhake and B. M. Bhanage, Catal. Commun., 2010, 11, 1233–1237 CrossRef CAS.
- B. Das, K. Venkateswarlu, A. Majhi, M. R. Reddy, K. N. Reddy, Y. K. Rao, K. Ravikumar and B. Sridhar, J. Mol. Catal. A: Chem., 2006, 246, 276–281 CrossRef CAS.
- Z. H. Zhang, L. Yin and Y. M. Wang, Adv. Synth. Catal., 2006, 348, 184–190 CrossRef CAS.
- E. Rafiee, H. Mahdavi, S. Eavani, M. Joshaghani and F. Shiri, Appl. Catal., A, 2009, 352, 202–207 CrossRef CAS.
- C. A. Brandt, A. C. M. P. da Silva, C. G. Pancote, C. L. Brito and M. A. B. da Silveira, Synthesis, 2004, 10, 1557–1559 CrossRef.
- M. Vaseem, A. Umar, Y. B. Hahn, D. H. Kim, K. S. Lee, J. S. Jang and J. S. Lee, Catal. Commun., 2008, 10, 11–16 CrossRef CAS.
- Y. L. Min, T. Wang and Y. C. Chen, Appl. Surf. Sci., 2010, 257, 132–137 CrossRef CAS.
- M. T. Qamar, M. Aslam, I. M. I. Ismail, N. Salah and A. Hameed, ACS Appl. Mater. Interfaces, 2015, 7, 8757–8769 CAS.
- Y. Xu, D. Chen, X. Jiao and K. Xue, J. Phys. Chem. C, 2007, 111, 16284–16289 CAS.
- D. P. Singh, A. K. Ojha and O. N. Srivastava, J. Phys. Chem. C, 2009, 113, 3409–3418 CAS.
- (a) S. Perrone, M. Capua, G. Cannazza, A. Salomone and L. Troisi, Tetrahedron Lett., 2016, 57, 1421–1424 CrossRef CAS; (b) Y. Liu, G. Wu and Y. Cui, Appl. Organomet. Chem., 2013, 27, 206–208 CrossRef CAS; (c) M. E. F. Braibante, H. S. Braibante, L. Missio and A. Andricopulo, Synthesis, 1994, 9, 898–900 CrossRef; (d) P. Davoli, I. Moretti, F. Prati and H. Alper, J. Org. Chem., 1999, 64, 518–521 CrossRef CAS.
- H. Zhang, Z. Wang, C. Wang, H. Wang, T. Cheng and L. Wang, RSC Adv., 2014, 4, 19512–19515 RSC.
- A. Kumar, L. Rout, R. S. Dhaka, S. L. Samala and P. Dash, RSC Adv., 2015, 5, 39193–39204 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, 1HNMR, and GC-MS data for the products. See DOI: 10.1039/c6ra22017g |
| This journal is © The Royal Society of Chemistry 2016 |