
Polyethyleneimine-stabilized hydroxyapatite nanoparticles modified with hyaluronic acid for targeted drug delivery†
Abstract
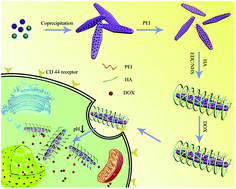
Targeted delivery of therapeutic drugs into cancer cells is a facile method to improve therapeutic efficacy. Focused on this point, a functionalized porous hydroxyapatite (HAp) nanoparticle-based nanocarrier consisting of polyethyleneimine (PEI) coating and outer hyaluronic acid (HA) modification (HAp–PEI–HA) has been developed. In this nanocarrier, PEI was coated onto the HAp to stabilize the nanoparticles, and HA acted as an efficient targeting ligand to selectively bind the CD44 receptors which are overexpressed on the surface of some cancer cells. Various techniques demonstrated the successful preparation of the HAp–PEI–HA nanoparticles. The resulting HAp–PEI–HA nanoparticles showed better dispersibility and stability in aqueous solution compared to bare HAp. In addition, the DOX released from DOX@HAp–PEI–HA exhibited a pH-responsive release profile. The targeting property of HAp–PEI–HA was investigated on A549 cells with high CD44 receptor expression and U87 cells with low CD44 receptor expression. The result showed enhanced cellular uptake of HAp–PEI–HA nanoparticles in A549 cells through CD44 receptor mediated cellular endocytosis. Furthermore, the effective targeted delivery of DOX by HAp–PEI–HA in A549 cells led to enhanced therapeutic efficacy. Thus, the designed HAp–PEI–HA nanocarrier showed promising potential for targeted drug delivery in cancer therapy.
 Please wait while we load your content...
Please wait while we load your content...

