
Synthesis and structure of color tunable and white-light emitting lanthanide metal–organic framework materials constructed from conjugated 1,1′-butadiynebenzene-3,3′,5,5′-tetracarboxylate ligand†
Jiewei Rongab,
Wenwei Zhang*a and
Junfeng Bai*a
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: wwzhang@nju.edu.cn; bjunfeng@nju.edu.cn
bSchool of Chemical and Materials Engineering, Huainan Normal University, Huainan, Anhui 232038, P. R. China. E-mail: zwj0076@126.com
First published on 24th October 2016
Abstract
A series of novel lanthanide metal–organic frameworks, namely [Ln2(BBTC)1.5(DMF)4]·2DMF·4H2O [BBTC4− = 1,1′-butadiynebenzene-3,3′,5,5′-tetracarboxylate, Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5), Er (6) and Yb (7)], have been prepared via solvothermal reaction. Single crystal X-ray analyses reveal that complexes 1–7 are isostructural and crystallize in the C2/c space group. They have the same three-dimensional (3D) architectures, topological analyses suggest that these frameworks possess the 3D (4,6)-connected network with Schläfli symbol of {43·63}3·{44·67·84}·{48·66·8}. Solid-state photoluminescent investigations at room temperature reveal that complex 2 and 4 emit the intensely red characteristic luminescence of Eu3+ ions and green luminescence emissions of Tb3+ ions, with a long lifetime up to 1.38 and 0.33 ms, respectively, during which the ligand emission of BBTC4− was quenched by the Eu3+ and Tb3+ ions. Complexes 5, 6 and 7 also display characteristic f–f luminescence emissions in the near-infrared region, respectively. These results indicate that BBTC4− is an ideal ligand with an “antenna effect” for Ln3+ ions, and an efficient energy transfer exists between them. Color tunable and white-light emitting materials were successfully achieved by carefully adjusting the doping concentration of Eu3+ and Tb3+ ions in the Gd3+ compound through trichromatic method.
Introduction
Recently, the design and synthesis of lanthanide metal–organic frameworks (Ln–MOFs), self-assembled from lanthanide metal ions and organic linkers, have attracted increasing attention in material research due to their superior functional properties and actual or potential applications.1–4 In comparison with transition metal ions, the lanthanide ions possess higher coordination numbers and more flexible coordination geometries, and thus lead to the formation of MOFs with versatile motifs.5 The lanthanide (4f) complexes usually show intense emission over narrow wavelength ranges and readily identifiable emission bands in both visible and NIR regions,6–8 which are potentially applicable as fluorescent probes and electroluminescent devices.9 However, lanthanide ions are very poor at absorbing light directly because of the low extinction coefficients of the Laporte forbidden f–f transitions, resulting in inefficient luminescence. Alternatively, the emission of the lanthanides can be sensitized by a coordinated ligand via “antenna effect”.10 In such a case, excitation occurs at the singlet energy state of a suitable organic ligand, which subsequently relaxes to its triplet state and then transfers energy to the lanthanide ions, and usually the conjugated organic multicarboxylate molecules are good candidates for “antenna” ligands of lanthanide ions.Up to now, Ln–MOFs have been extensively studied in sensing ions, vapors, explosives, etc.11 And recently, tunable white-light emission of Ln3+ complexes has also been reported since white-light emitting materials and devices have potential applications in general lighting, low-cost back-lighting and fullcolor displays.12,13 Qian et al. have reported a rational design strategy to construct white-light emitting materials by doping Eu3+ and Tb3+ ions into isostructural La3+ ions metal–organic framework.14 Zheng, Li and Zang et al. also achieved strong white-light emitters by varying the stoichiometric ratio of Eu3+, Tb3+ and La3+/Ga3+ ions within the mixed isostructural lanthanide metal–organic complexes.15 Moreover, single-phase Eu–Ag dual component white-light emitters have also been reported through assembly of d–f heterometallic MOFs.16 Generally, the white-light emitting materials are mainly produced through monochromatic, dichromatic, trichromatic, and tetrachromatic approaches.13 Among them, the solid state trichromatic white-light emitting materials have attracted much more attention in terms of their good color rendering properties and high luminous efficiency, and white-light emitting Ln–MOFs are candidates for light-emitting devices due to their structural diversity and ligand-dependent luminescence sensitization. But it is still very challenging and difficult to target white-light emitting Ln–MOF materials because different light emitters should compensate exactly through the trichromatic approaches. As is known, the Gd3+ complex sometimes may act as a blue emitter and shows ligand-centered visible emission in blue light region owing to the higher energy (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 150 cm−1) of the Gd3+ lowest emitting level, and Eu3+ and Tb3+ complexes usually emit the intensely red characteristic luminescence of Eu3+ ions and green luminescence of Tb3+ ions. Therefore, Gd3+, Eu3+, and Tb3+ complexes might be incorporated into the resulting trichromatic white-light emitting materials.
150 cm−1) of the Gd3+ lowest emitting level, and Eu3+ and Tb3+ complexes usually emit the intensely red characteristic luminescence of Eu3+ ions and green luminescence of Tb3+ ions. Therefore, Gd3+, Eu3+, and Tb3+ complexes might be incorporated into the resulting trichromatic white-light emitting materials.
Recently, our group and others have focused on developing and designing π-conjugated ligands with alkyne functionality, such as H4EBTC (EBTC4− = 1,1′-ethynebenzene-3,3′,5,5′-tetracarboxylate), H4BBTC (BBTC4− = 1,1′-butadiynebenzene-3,3′,5,5′-tetracarboxylate) and H3CPEIP (CPEIP3− = 5-[(4-carboxyphenyl)ethynyl]isophthalic acid).17,18 The study on transition metal MOFs revealed that they are good ligands to construct porous MOFs with significant uptake of gases (H2, CO2, N2, CH4 or C2H2), especially for acetylene due to the incorporation of the triple C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond into the organic backbone, and the investigation on lanthanide MOFs based on them indicated that they are efficient “antenna effect” ligands for enhancing Eu3+ or Tb3+ emission of f–f transitions. Even so, as far as we know, the systematic study on the Ln–MOFs' construction and property based on them are less reported, especially for H4BBTC, and no work has been covered on white-light emitting materials of Ln–BBTC MOFs until now, thus we selected H4BBTC as a ligand to design and synthesize Ln–BBTC MOFs so as to systematically investigate their photoluminescent properties and try to obtain color tunable and white-light emitting materials. Moreover, compared with EBTC4−, BBTC4− is more flexible since the butadiyne group can make the two aromatic rings rotate more freely than those linked by the ethyne group, which may contribute to constructing MOFs with versatile motifs, and it has both aromatic ring and linear polyyne chain functionalities, which may potentially serve as specific sites for certain molecular recognition, unique gas sorption, or particular catalysis applications. All above considerations inspired us to explore new Ln–MOFs with rectangular-planar 4-connected tetracarboxylates ligand of H4BBTC. In this contribution, we synthesized seven new Ln–MOFs [Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5), Er (6) and Yb (7)] based on it and systematically studied their crystal structures and photoluminescent properties. Moreover, we also prepared series of MOFs-based co-doped materials EuxGd1−x–BBTC, TbyGd1−y–BBTC and EuxTbyGd1−x−y–BBTC, and investigated the luminescence natures of them. And ultimately, we successfully obtained tunable white-light emitting materials through careful adjustment of the relative concentration of the doping of Eu3+ and Tb3+ ions in the Gd3+ complex.
C bond into the organic backbone, and the investigation on lanthanide MOFs based on them indicated that they are efficient “antenna effect” ligands for enhancing Eu3+ or Tb3+ emission of f–f transitions. Even so, as far as we know, the systematic study on the Ln–MOFs' construction and property based on them are less reported, especially for H4BBTC, and no work has been covered on white-light emitting materials of Ln–BBTC MOFs until now, thus we selected H4BBTC as a ligand to design and synthesize Ln–BBTC MOFs so as to systematically investigate their photoluminescent properties and try to obtain color tunable and white-light emitting materials. Moreover, compared with EBTC4−, BBTC4− is more flexible since the butadiyne group can make the two aromatic rings rotate more freely than those linked by the ethyne group, which may contribute to constructing MOFs with versatile motifs, and it has both aromatic ring and linear polyyne chain functionalities, which may potentially serve as specific sites for certain molecular recognition, unique gas sorption, or particular catalysis applications. All above considerations inspired us to explore new Ln–MOFs with rectangular-planar 4-connected tetracarboxylates ligand of H4BBTC. In this contribution, we synthesized seven new Ln–MOFs [Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5), Er (6) and Yb (7)] based on it and systematically studied their crystal structures and photoluminescent properties. Moreover, we also prepared series of MOFs-based co-doped materials EuxGd1−x–BBTC, TbyGd1−y–BBTC and EuxTbyGd1−x−y–BBTC, and investigated the luminescence natures of them. And ultimately, we successfully obtained tunable white-light emitting materials through careful adjustment of the relative concentration of the doping of Eu3+ and Tb3+ ions in the Gd3+ complex.
Experimental section
Chemicals and reagents
1,1′-Butadiynebenzene-3,3′,5,5′-tetracarboxylate (H4BBTC) was synthesized according to the method published before.18a LnCl3·nH2O (Ln = Eu, Tb, Dy, Er and Yb) were prepared by the reaction of hydrochloric acid (6.0 M) with the corresponding lanthanide oxide, and then were evaporated at 80 °C. SmCl3·6H2O and GdCl3·6H2O were purchased from chemical company and used directly. N,N-Dimethyl formamide (DMF) was dried and distilled according to standard procedures. All commercially available chemicals were of analytical grade and used without further purification.Physical measurements
Powder X-ray diffraction (PXRD) measurements were performed on a Bruker D8 Discover diffractometer with Cu Kα (λ = 1.54056 Å) radiation with a scan speed of 5° min−1 and a step size of 0.02° in 2θ at room temperature. Elemental analyses for C, H and N were carried out on a PerkinElmer 240 elemental analyzer. Inductively coupled plasma (ICP) was performed on a PerkinElmer Optima 5300DV spectrometer. The morphology, elemental mapping images and composition of the samples were inspected using a scanning electron microscope (SEM, S-3400N II and S-4800, Hitachi) equipped with an energy-dispersive X-ray spectroscope (EDX, EX-250 and S 4800, HORIBA). The IR spectra were recorded on a NICOLET iS10 spectrometer using KBr pellets. Thermal gravimetric analyses (TGA) were measured using a DTA-TGA 2960 thermogravimetric analyzer with a heating rate of 20 °C min−1 under nitrogen atmosphere. The UV/visible absorbance in the solid state at room temperature was performed on Shimadzu UV-3600 spectrometer with BaSO4 as reflectance standard. The photoluminescence spectra in the solid state were recorded by F-7000 FL and RF-5301 PC spectrophotometer at room temperature, respectively. The luminescence decay curves were measured by an Edinburgh Instruments FLS920P spectrometer equipped with Xe900 xenon lamp.Preparation and characterization of Ln–MOFs
The analogous procedure above-mentioned was used for preparation of 2–7 (see ESI†), just replaced SmCl3·6H2O by other LnCl3·6H2O. The microanalysis and main IR bands are summarized in Table S1.† Moreover, the doped complexes EuxGd1−x–BBTC, TbyGd1−y–BBTC and EuxTbyGd1−x−y–BBTC were prepared by adopting the analogical method as for the isostructural Ln analogies, loading the corresponding LnCl3·6H2O (Ln3+ = Gd3+, Eu3+ or Tb3+) as the starting materials in different stoichiometric ratios. Their purities were confirmed by powder XRD (Fig. S1 and S2†) and elemental analysis of C, H and N (Tables S2 and S3†), energy dispersive X-ray (EDX) elemental mapping analysis for Eu0.25Gd0.75–BBTC, Tb0.5Gd0.5–BBTC and Eu0.045Tb0.055Gd0.9–BBTC prove that each EuxGd1−x–BBTC/TbyGd1−y–BBTC/EuxTbyGd1−x−y–BBTC doped complex is not a simple mixture but a single phase coordination polymer (Fig. S3†), and inductively coupled plasma (ICP) analysis of these co-doped compounds further confirms that the amounts of Gd3+, Eu3+ and/or Tb3+ ions in them are almost identical with the concentration of the starting materials (Table S4†).
Crystallographic analyses
Single crystal X-ray analyses were conducted on a Bruker Smart Apex II CCD diffractometer using graphite monochromated Mo/Kα radiation (λ = 0.71073 Å) at 296 K. Data reductions and absorption corrections were performed using the SAINT and SADABS software packages,19 respectively. The structures were solved by direct methods with the SHELXL-97 software package.20 All hydrogen atoms were placed at the calculated positions and refined riding on the parent atoms. All non-hydrogen atoms were refined anisotropically using the full-matrix least-squares method on F2. In complex 1–7, solvent molecules in the structure were highly disordered and were impossible to refine using conventional discrete-atom models. To resolve this issue, the contribution of solvent electron density was removed by SQUEEZE routine in PLATON.21 It should be noted that the weak diffraction intensity of 1 and 3 with small single crystals result in the data with low completeness, which are shown in Table S5.† The main data of collection and refinement details of complexes 2, 4, 5, 6 and 7 are given in Table 1. Selected bond lengths and angles are listed in Tables S6 and S7.† The CCDC reference numbers are 1478788 for 2, 1478790 for 4, 1478791 for 5, 1478792 for 6 and 1478793 for 7, respectively.| a R1 = Σ||F0| − |Fc||/Σ|F0|, wR2 = {Σ[w(F02 − Fc2)2]/Σ[w(F02)2]}1/2. | |||||
|---|---|---|---|---|---|
| Compound | 2 | 4 | 5 | 6 | 7 |
| Formula | C48H59Eu2N6O22 | C48H59Tb2N6O22 | C48H59Dy2N6O22 | C48H59Er2N6O22 | C48H59Yb2N6O22 |
| Formula weight | 1376 | 1390 | 1397 | 1407 | 1418 |
| Temperature (K) | 296(2) | 296(2) | 296(2) | 296(2) | 296(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c | C2/c | C2/c | C2/c |
| a (Å) | 30.0058(13) | 29.9342(11) | 29.9537(12) | 29.9258(10) | 29.8541(10) |
| b (Å) | 21.2440(9) | 21.1600(7) | 21.1398(9) | 21.0418(7) | 20.9459(6) |
| c (Å) | 19.5943(9) | 19.5148(7) | 19.4385(8) | 19.4326(6) | 19.3789(6) |
| α (deg) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| β (deg) | 93.870(1) | 93.9350(1) | 93.980(1) | 94.000(1) | 94.071(1) |
| γ (deg) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| V (Å3) | 12461.8(9) |
12331.7(8) |
12279.1(9) |
12206.8(7) |
12087.5(7) |
| Z | 8 | 8 | 8 | 8 | 8 |
| F(000) | 4568 | 4600 | 4616 | 4648 | 4680 |
| GOF | 1.070 | 1.033 | 1.013 | 1.082 | 1.099 |
| R1, wR2 [I > 2σ(I)]a | 0.0595, 0.1998 | 0.0508, 0.1564 | 0.0684, 0.2140 | 0.0660, 0.2122 | 0.0618, 0.1951 |
Results and discussion
Crystal structures
Compounds 1–7 were obtained under the same conditions by the mild solvothermal reactions. Single crystal X-ray diffraction analyses reveal that all the compounds are isomorphous, crystallize in the monoclinic C2/c space group and exhibit a 3D framework. Powder X-ray diffraction (PXRD) of compounds 1–7 were recorded at room temperature. As shown in Fig. 1 and S4,† the peak positions of the theoretical and experimental PXRD patterns are in good agreement with each other, which clearly indicate the high purity and homogeneity of these samples. Besides this, the IR spectra of 1–7 are almost identical (Fig. S5†), and the microanalysis disclosed that complexes 1–7 have the analogous chemical components with a formula of [Ln2(BBTC)1.5(DMF)4]·2DMF·4H2O [Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5), Er (6) and Yb (7)]. In addition, the doped compounds EuxGd1−x–BBTC, TbyGd1−y–BBTC and EuxTbyGd1−x−y–BBTC are also isostructural with the parent complex 3, which are verified by powder X-ray diffraction (PXRD) analysis (Fig. S1 and S2†), as a result, only the structure of [Tb2(BBTC)1.5(DMF)4]·2DMF·4H2O (4) is representatively described in detail. The structures of 1, 2, 3, 5, 6 and 7 are shown in Fig. S6.†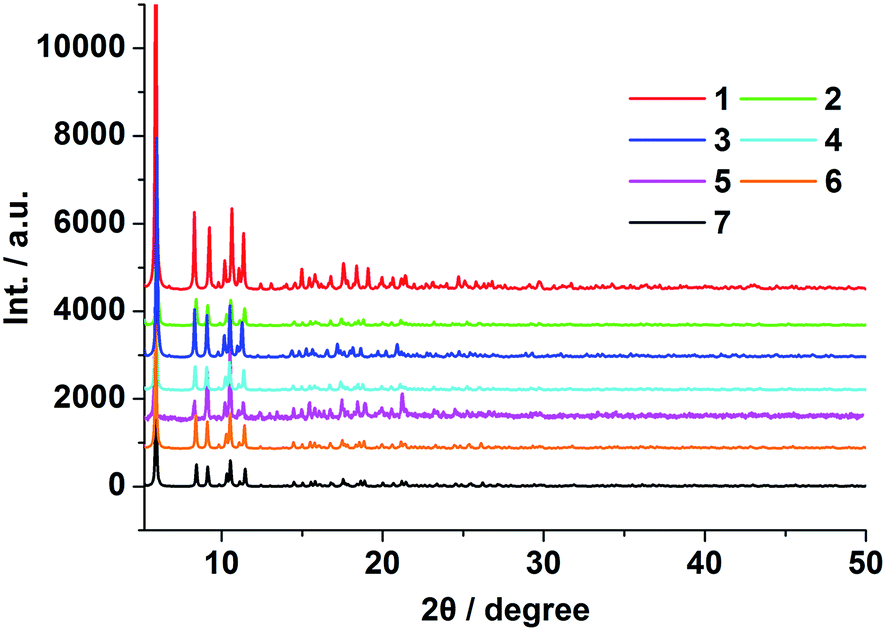 | ||
| Fig. 1 PXRD patterns of 1–7 at ambient temperature. | ||
Crystal 4 belongs to the monoclinic system with space group of C2/c. As shown in Fig. 2a, its asymmetric unit cell contains two eight-coordinated Tb3+ ions, one and a half BBTC4− and four coordinated DMF molecules. Two crystallographically independent Tb3+ ions (labeled as Tb1 and Tb2, respectively) are coordinated with eight oxygen atoms, exhibiting two different coordination geometries (Fig. 2b). The bond lengths of Tb–O bond distances range from 2.277(5) to 2.471(4) Å (Table S6†), and the O–Tb–O bond angles span from 48.6(7) to 156.6(6)° (Table S7†). These bond parameters are similar to the values of Ln–carboxylate complexes in literatures.22 The coordination geometry around Tb1 ions can be described as a distorted bicapped trigonal prism with its eight coordinated oxygen atoms coming from five BBTC4− ligands (O1, O2, O7a, O8b, O11 and O12c) and two DMF molecules (O17 and O19). Among the five coordinated BBTC4− ligands, one carboxylate from one BBTC4− coordinates to Tb1 as a chelating mode, while the other four carboxylates from four different BBTC4− ligands coordinate to two Tb1 centers in a bidentate bridging coordination mode to give a dinuclear Tb2(CO2)4 (A) subunit (SBU) with Tb1⋯Tb1 distance of 5.537 Å within this SBU (Fig. 2c). As for Tb2 ions, they bond to six oxygen (O3, O4d, O5e, O6e, O9 and O10) from four BBTC4− ligands and the other two oxygen (O18 and O20) from two DMF molecules, resulting in distorted trigonal dodecahedral geometries. Similarly, two Tb2 centers are linked by two carboxylates in a bidentate bridging coordination mode from two different BBTC4− ligands to form a binuclear Tb2(CO2)2 (B) subunit (SBU), with Tb2⋯Tb2 distance of 5.480 Å. Furthermore, two more carboxylates from another two BBTC4− ligands coordinate to Tb2 as chelating mode. Ultimately, two kinds of BBTC4− ligand (α and β form) with different steric configurations generate upon complexation. As shown in Fig. 2c, the torsion angle of the butadiyne group (C29–C30–C30j–C29j) is 4.289° and the dihedral angle between the two aromatic rings is 1.467° in α form, while those in β form are 6.427° (C9–C10–C11–C12) and 88.447°, respectively. That's to say, compared with EBTC4− in constructing Ln–MOFs,18b BBTC4− is more flexible since the longer butadiyne chain can make the two aromatic rings rotate more freely than those linked by the ethyne group. As a result, the butadiyne chain in BBTC4− is bent to two different degree, with one-half of the aromatic rings of the ligands rotated round it to be almost perpendicularly arranged so as to satisfy the coordination geometry of Tb3+ (β form). Moreover, in the α-BBTC4− ligand, two carboxylates in the cis-positions adopt bidentate mode to bridge two Tb1 centers, and the other two adopt chelating mode to bind two Tb2 atoms. While in the β-BBTC4− ligand, two opposite carboxylates in two phenyl rings adopt a bidentate bridging mode to link two Tb1 and two Tb2 ions, and the other two also in the para-positions adopt a chelating mode to coordinate one Tb1 and one Tb2 ion, respectively. Overall, each Tb2(CO2)4 (A) and Tb2(CO2)2 (B) SBU is surrounded by six BBTC4− bridging ligands and each BBTC4− ligand connects two Tb2(CO2)4 (A) and two Tb2(CO2)2 (B) SBUs, generating a three-dimensional (3-D) open framework. As shown in Fig. 2f, the coordinated DMF molecules, being disordered, locate in the channels, and there exist three types of one-dimensional channels along the c axis, which possess the open window of 10.3 × 6.3 Å (C), 7.9 × 5.8 Å (D) and 10.1 × 8.6 Å (E) when the van der Waals radii of the atoms at the wall of the channel are subtracted, respectively. Moreover, open channel systems also exist along the a and b axis, with an open window size of 6.7 × 6.3 Å and 6.3 × 3.9 Å, respectively (Fig. 2d and e). The total potential void volume is 63.4% calculated from PLATON/SOLV21 if the guest water, guest DMF and coordinated DMF molecules are removed (Fig. S7†). From the view point of topology, the 3-D structure of 4 can be described as a (4,6)-connected network with Schläfli symbol of {43·63}3·{44·67·84}·{48·66·8} by considering each Tb2(CO2)4 (A) and Tb2(CO2)2 (B) SBU as a 6-connected node and the organic ligand as a 4-connected node (Fig. 2g).
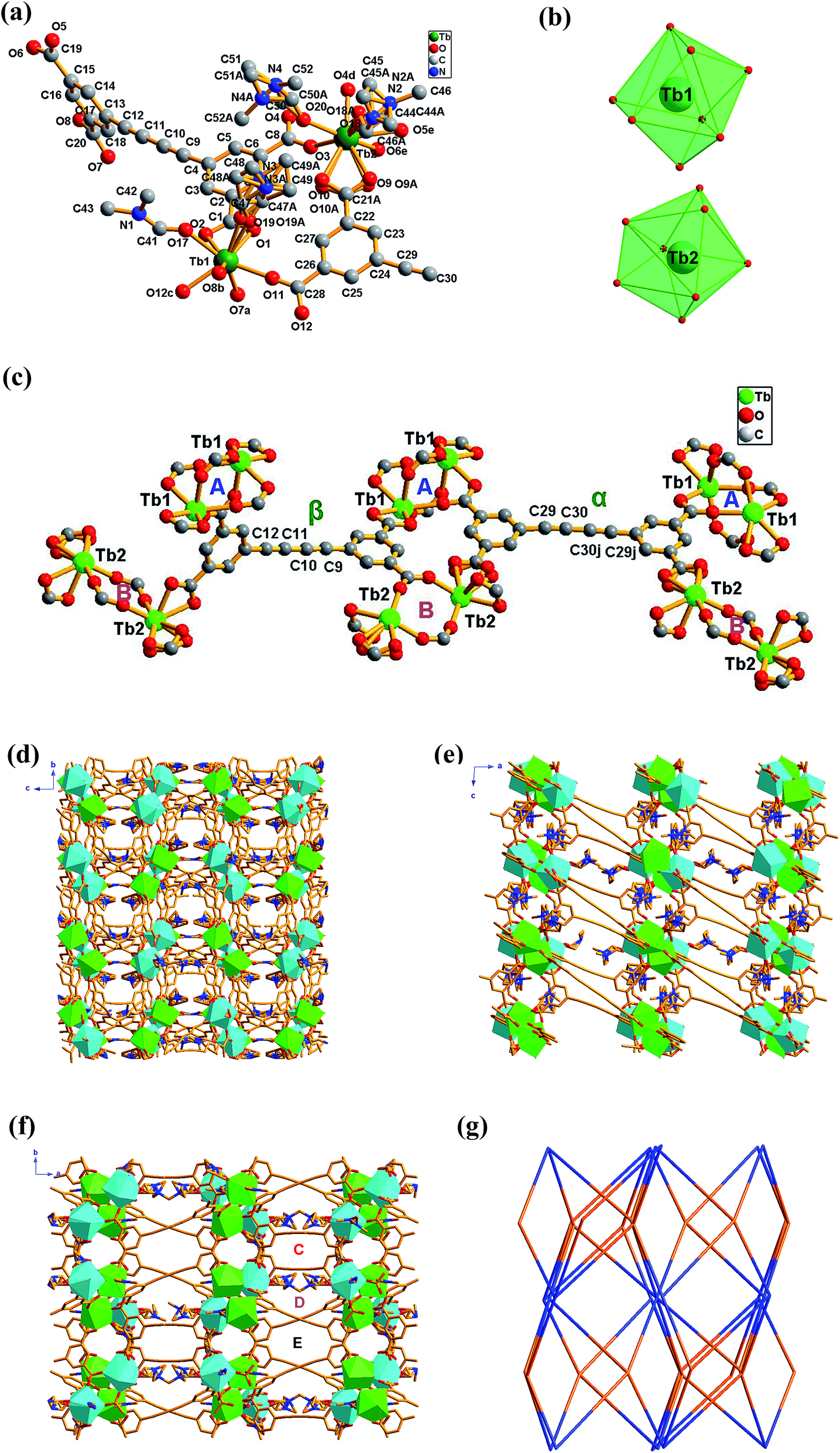 | ||
| Fig. 2 (a) Coordination environments of Tb3+ ions with the H atoms omitted for clarity; symmetry codes: a = −x, −y, 1 − z; b = 0.5 + x, 0.5 + y, z; c = 0.5 − x, 0.5 − y, 1 − z; d = 0.5 − x, −0.5 − y, 1 − z; e = 0.5 + x, −0.5 − y, 0.5 + z; j = 1 − x, 1 + y, 1.5 − z; (b) coordination polyhedron of Tb3+ ions; (c) the local coordination environment of the Tb1 and Tb2 ions in 4. A and B represent Tb2(CO2)4 and Tb2(CO2)2 subunits, respectively; α and β represent BBTC4− ligands with two different steric forms; (d–f) polyhedral representation of 4 seen from a, b, and c directions (the green and turquoise polyhedra correspond to [Tb1O8] and [Tb2O8] units, respectively); (g) (4,6)-connected network presented, orange represents the 4-connected node of organic ligand and blue represents the 6-connected node of SBU in 4. | ||
It should be noted that the structures of Ln–BBTC MOFs in this work have some difference compared with our previous reported Eu–MOFs constructed from the same ligand, such as the torsion angle of the butadiyne group and the dihedral angle between the two aromatic rings in BBTC4−, even if their coordination modes of the ligand are similar (bidentate bridging and chelating mode). In Ln–BBTC of this work, the smaller torsion angle of the butadiyne group in both α and β form (4.289° and 6.427°) exhibits that the butadiyne has better linearity (the torsion angle of the butadiyne group in α and β form is 150.973° and 14.073° respectively in previous reported Eu–MOFs18a); moreover, the smaller dihedral angle between the two aromatic rings in α form (1.467°) and the larger one in β form (88.447°) suggest that the two aromatic rings have better coplanarity or perpendicularity (the dihedral angle between the two aromatic rings in α and β form is 8.708° and 87.340° respectively in Eu–MOFs18a). The different steric configuration of BBTC4− ligand between Ln–BBTC and Eu–MOFs should be mainly ascribed to the different coordination environment of the lanthanide ions, since they have different coordinating solvents besides BBTC4− ligand. And the different synthetic conditions, such as solvent and temperature, lead to the different coordination environment.
TG analyses
TGA measurements of compounds 1–7 were performed from room temperature to 750 °C under a nitrogen atmosphere to study the thermal stability of these lanthanide metal–organic frameworks. As shown in Fig. S8,† the isomorphism 1–7 exhibit similar thermal behavior with several continuous weight loss processes below 354 °C, and these weight loss processes correspond to the guest water, guest DMF molecules and coordinated DMF molecules releasing. As a representative example, compound 1 was discussed in detail. From the TG curve depicted in Fig. 3, compound 1 shows two main steps of sequential weight loss process with a percentage of 15.88% before 162 °C, corresponding to the release of four guest water molecules and two guest DMF molecules (calcd: 15.89%), and 21.29% in 162–354 °C temperature range, attributing to the loss of four coordinated DMF molecules (calcd: 21.27%). Above 354 °C, the whole framework gradually begins to break down upon further heating.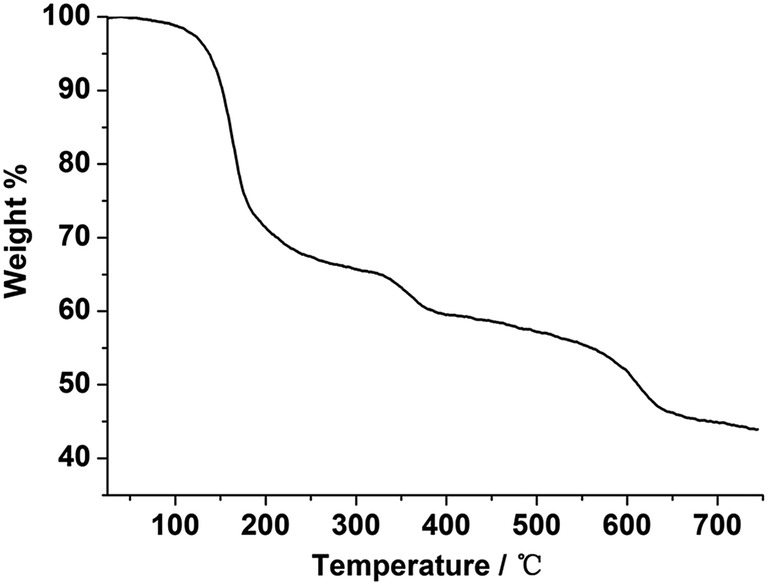 | ||
| Fig. 3 TG curve of 1. | ||
Photoluminescence properties
To investigate the luminescence properties of the lanthanide metal–organic frameworks, the UV-vis absorption spectrum for H4BBTC was recorded in the solid state at room temperature (Fig. S9(a)†). It is observed that H4BBTC exhibits intense broad bands centered at 340 nm, which can be attributed to π–π* transitions of the aromatic rings. And consequently, it gives a broad emission centered at 420 nm upon excitation at 340 nm due to π ← π* transitions (Fig. S9(b)†).The solid-state luminescence properties of complexes 1–7 were investigated at room temperature. Upon excitation at 340 nm, compound 3 displays strong blue luminescence (ref. the CIE chromaticity diagram Fig. 6b) with a single broad emission band centered at 467 nm (Fig. 4, its excitation spectrum is given in Fig. S10(b)†), such a broad band emission can also be assigned to the intraligand π ← π* transition of BBTC4−. In comparison to the emission of H4BBTC, compound 3 obviously shifts towards the lower energy side (red-shift), which may be attributed to the fact that the lowest excited state of Gd3+ (6P7/2) is too high to accept energy from the ligand, making its characteristic 4f–4f transition invisible.23,24 Therefore, the red-shift of the intense emission in 3 could be attributed to the coordination effect, which affects the HOMO and LUMO levels of the ligand to change the transition energy, and enables the rigidity of the aromatic backbones and reduces the loss of energy by radiationless decay of the intraligand emissions.25
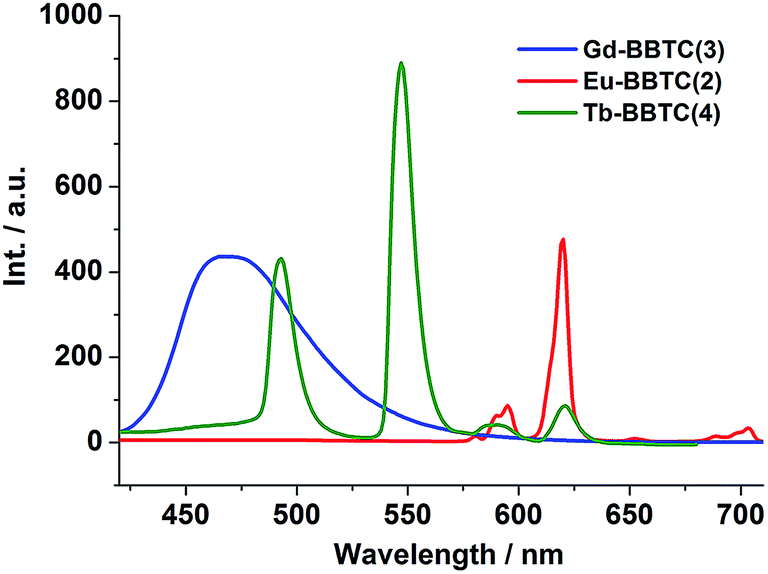 | ||
| Fig. 4 Emission spectra of 2, 3 and 4 under excitation λex = 340 nm at room temperature. | ||
Compound 1, excited at 340 nm, displays characteristic orange luminescence of Sm3+ ions in the visible region (ref. the CIE chromaticity diagram Fig. 6b), and the typical emission bands centered at 563, 599 and 646 nm (Fig. S11(a)†) are assigned to the transitions from 4G5/2 to 6HJ (J = 5/2, 7/2 and 9/2), respectively.
As shown in Fig. 4, compound 2 shows five characteristic emission bands of Eu3+ ions centered at 579, 592, 615, 653 and 701 nm with red luminescence upon excitation at 340 nm (ref. the CIE chromaticity diagram Fig. 6b). Five typical emission bands are corresponding to the 5D0 → 7FJ (J = 0–4) transitions, respectively (its excitation spectrum is given in Fig. S10(a)†). The symmetric forbidden emission 5D0 → 7F0 at 579 nm is a weak band. The emission at 592 nm is corresponding to the magnetic dipole induced 5D0 → 7F1 transition, which is fairly insensitive to the environment of the Eu3+ ions, while the most intense emission at 615 nm is attributed to the electric dipole induced 5D0 → 7F2 transition, which is hypersensitive to the coordination environment of the Eu3+ ions. Moreover, the peak around 653 nm for the 5D0 → 7F3 transition is observed with weak intensity, and the electric dipole 5D0 → 7F4 transition around 701 nm is comprised of two peaks. It is well known that the intensity ratio between 5D0 → 7F2 and 5D0 → 7F1 transitions is strongly relevant to the local symmetry of the Eu3+ ions. Specifically, the transition intensity of 5D0 → 7F2 increases significantly as the site symmetry of the Eu3+ ions are lowered, whereas the local coordination environment of Eu3+ ions have no clear impact on the 5D0 → 7F1 transition.26 As for 2, the intensity ratio of I(5D0 → 7F2)/I(5D0 → 7F1) is ca. 5, further indicating that the symmetry of Eu3+ ions is low and the Eu3+ ions are not located at the inversion center, which is in good agreement with our X-ray structural analyses. It is worth noting that the characteristic emission peaks from BBTC4− ligand disappeared, which indicates the effective sensitization of Eu3+ ions from ligand excitation.
Upon sensitization of the Tb3+ ion by means of ligand excitation, compound 4 displays intense green luminescence when excited at 340 nm (ref. the CIE chromaticity diagram Fig. 6b), exhibiting four emission bands centered at 487, 542, 581 and 618 nm which are attributed to the 5D4 → 7FJ (J = 6, 5, 4, 3) transitions, respectively (Fig. 4, its excitation spectrum is shown in Fig. S10(c)†).27 As expected, the 5D4 → 7F5 (sensitive to the nature of the surrounding atoms) is the strongest one according to its largest probability for both electric-dipole and magnetic-dipole-induced transitions.28 Similar to that of compound 2, no ligand-based emission is observed, thereby also suggesting effective ligand to metal energy transfer.
When excited at 340 nm at room temperature, compound 5 displays photoluminescent behavior with typical Dy3+ emissions centered at 483 and 576 nm (Fig. S11(b),† its excitation spectrum is given in Fig. S10(d)†), which correspond to the characteristic emission of 4F9/2 → 6HJ (J = 15/2 and 13/2) transitions of Dy3+ ions respectively.
We have also investigated the emission in the near-IR region of compound 5, which is a rarely described phenomenon.29 Three emission bands centered at 950 (4F9/2 → 6F7/2), 976 (4F9/2 → 6F5/2) and 1147 nm (4F9/2 → 6F3/2) are observed for the Dy3+ complex (Fig. S11(c)†). Notably, the emission of Dy3+ complex is not a single sharp transition with the presence of well-split NIR emission peaks, which may be ascribed to the crystal-field splitting.30
Upon excitation at 340 nm, compound 6 also gives rise to the characteristic NIR emission (Fig. S11(d)†). The emission band centered at 1533 nm covers large spectral range extending from 1450 to 1652 nm, ascribing to the typical 4I13/2 → 4I15/2 transition of Er3+ ions. Erbium-doped materials have been attracted much interest for many years, because the transition around 1540 nm is in the right position of the third telecommunication window.31
Compound 7 shows a broad band in the range of 920–1100 nm, comprised of a sharp peak at 977 nm when excited at 340 nm (Fig. S11(e)†), which is attributed to the 2F5/2 → 2F7/2 transition and broader vibronic components at the longer wavelength.32 The NIR emission of 7 is not a single sharp band but a broad band, this should be caused by the splitting of the energy levels of the Yb3+ ions as a consequence of ligand field effect.33
The luminescence decay curves of 1–7 were obtained in the solid state at room temperature (Fig. 5 and S12†). The decay curves are well fitted into a single exponential function: y = y0 + A1 × exp(−t/τ), where y0 and A1 are two constants, and τ is the decay constant defined as the photoluminescence lifetime. The main emission peaks at 599, 615, 467, 542, 576, 1533 and 977 nm were monitored, respectively. The black dots are the raw experimental data and the red lines represent the fitting curves. The best fit of the experimental photoluminescence intensities to the above equation led to the following photoluminescence lifetimes: τ = 1.68 μs for 1, 1.38 ms for 2, 2.36 μs for 3, 0.33 ms for 4, 1.86 μs for 5, 3.34 μs for 6 and 2.91 μs for 7, respectively. In general, the f–f electronic transitions of lanthanide ions are forbidden, leading to long excited state decay time. In this work, it was found that the photoluminescence lifetimes of 2 and 4 are at millisecond magnitude order, falling in the range of lanthanide ion luminescence decay time 10–2000 μs.34
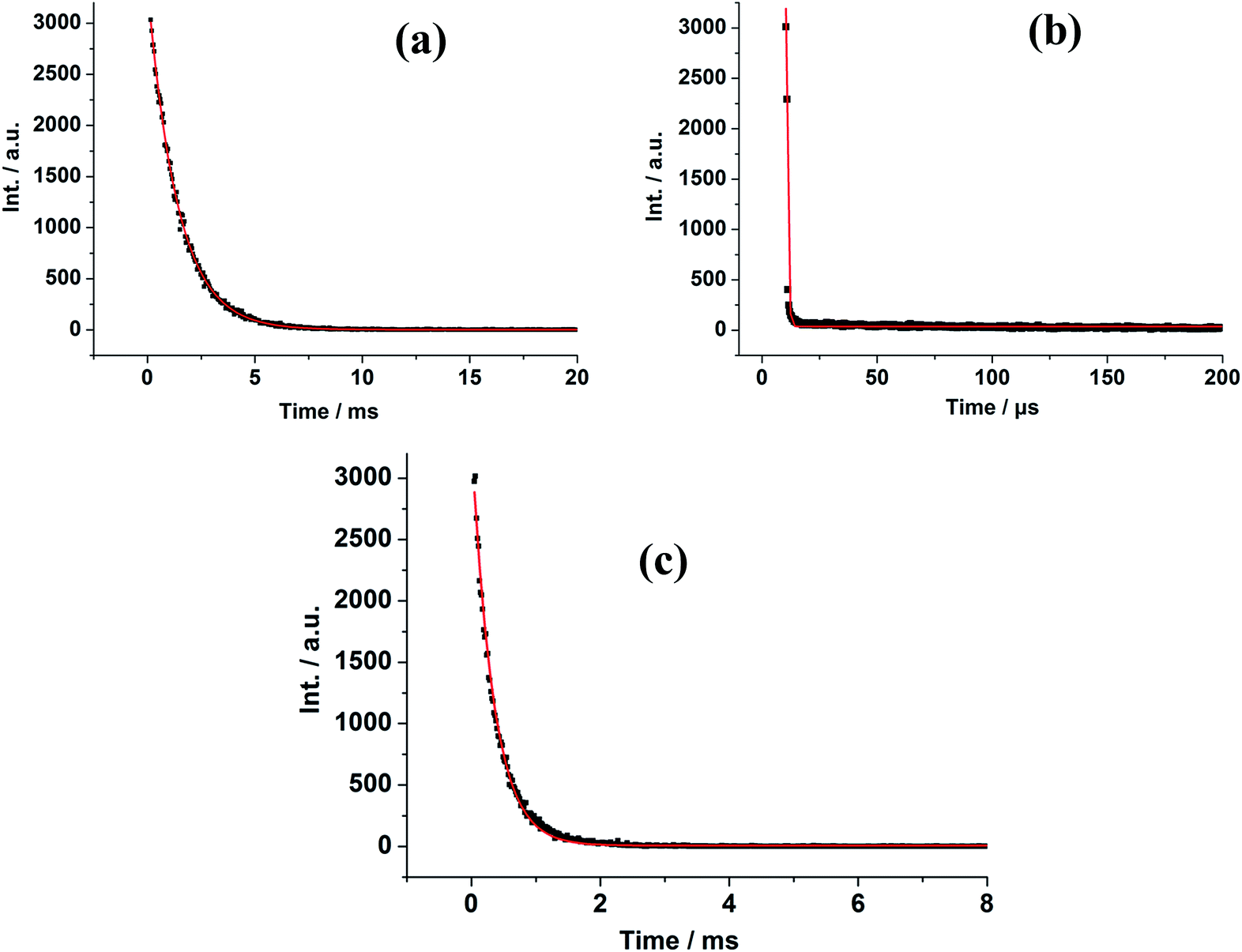 | ||
| Fig. 5 Luminescence decay profiles for 2 (a), 3 (b) and 4 (c). | ||
Since the whole family are isostructural, it is expected that we can easily tune their emissions by doping methods. Among the isostructural Ln–BBTC MOFs, Eu–BBTC (2), Tb–BBTC (4) and Gd–BBTC (3) emitted intense red, green and blue emissions, respectively (Fig. 4), therefore, we engaged in the preparation of monodoped EuxGd1−x–BBTC and TbyGd1−y–BBTC compounds, and bidoped EuxTbyGd1−x−y–BBTC compounds, to investigate whether the different percent compositions would result in different colors and the white-light emission would be successfully achieved due to photoluminescence properties of the co-doped MOFs.
The solid-state emission spectra of the monodoped EuxGd1−x–BBTC and TbyGd1−y–BBTC compounds were investigated upon excitation at 340 nm (Fig. S13(a) and S14(a),† the solid-state excitation spectra of Eu0.25Gd0.75–BBTC and Tb0.5Gd0.5–BBTC are shown in Fig. S15(a) and (b)†). As expected, both exhibited characteristic emissions of Eu3+ and Tb3+ ions, or ligand-to-metal charge transfer, and along with the increase of Eu3+ and Tb3+ ions concentration, the luminescence intensity corresponding to the characteristic Eu3+ (615 nm) and Tb3+ (542 nm) ions almost linearly increased, while the luminescence intensity from BBTC4− ligand (467 nm) slightly decreased, which could be stemmed by Ln3+ ions concentration effects.35 It is of note that the Eu3+- or Tb3+-based emissions are systematically more intense than that of the BBTC4− ligand in these monodoped compounds (Fig. S13(a) and S14(a)†), suggesting that partial energy transfers from Gd3+ to Eu3+ or Tb3+ ions and that the Ln3+-centered luminescence is significantly sensitized by the π-conjugated organic multicarboxylate ligands.15a As a result, tunable colors could be readily generated from blue to pale pink, pale orange and red for EuxGd1−x–BBTC compounds and from blue to pale blue and green for TbyGd1−y–BBTC compounds, respectively (Fig. S13(b) and S14(b)†). The corresponding CIE chromaticity coordinates of these monodoped complexes are listed in Tables S8 and S9.†
The tunable colors from monodoped EuxGd1−x–BBTC and TbyGd1−y–BBTC compounds prompt us to investigate the photoluminescence properties of the bidoped EuxTbyGd1−x−y–BBTC compounds by compensating the red color from Eu3+ ions, green color from Tb3+ ions and blue color from BBTC4− ligands so as to obtain the trichromatic white-light emission materials by adjusting the relative concentration of Eu3+, Tb3+ and Gd3+ ions in the co-doped EuxTbyGd1−x−y–BBTC MOFs. This would be of interest since white-light emitting materials have extensive applications in general lighting sources. Since white-light emission can be produced when the emission intensities around red, green and blue range are comparable,15,36 and the energy transfer should be incomplete from the Gd3+ host to doped Ln3+ ions, which is the most significant feature to realize the white-light emission in the co-doped system.15c We made an effort to optimize the relative concentration of Ln3+ dopants in EuxTbyGd1−x−y–BBTC MOFs, and the emission spectra of the doped compounds (A–E) which exhibit the typical 5D0 → 7F2 transition around 615 nm for Eu3+ in 2, 5D4 → 7F5 transition around 542 nm for Tb3+ in 4, and the broad band centered at 467 nm assigned to intraligand charge transfer of the ligands in 3 were obtained upon excitation at 340 nm (Fig. 6a, the solid-state excitation spectra of Eu0.015Tb0.035Gd0.95–BBTC is given in Fig. S15(c)†). We can also see that the luminescence intensity of the Eu3+ and Tb3+ ions decreased significantly as the concentration of Eu3+ and Tb3+ ions was decreased due to the concentration effects (A–E),35 while the emission intensity resulting from BBTC4− ligands increased gradually, thus combination of the emissions from f–f transitions of Eu3+ and Tb3+ ions and those from BBTC4− ligands can readily generate variations in the relative red, green and blue emission intensities when the doping ratios of Ln3+ dopants were changed, resulting in tunable colors (A–E) from pale yellow to pale yellow-green, pale green, near white, and white (Fig. 6b). The corresponding CIE chromaticity coordinates of these doped complexes are listed in Table S10.† All above reveals that the emission color of EuxTbyGd1−x−y–BBTC MOFs can be finely tuned through different combination of the amount of Ln3+ dopants, and ultimately, the white-light emission is readily produced when the red emission of Eu3+ ions, the green emission of Tb3+ ions, and the blue emission of BBTC4− ligands occur simultaneously, and the emission peaks at 615, 542 and 467 nm are comparable in intensity (Fig. 6 and S16†), with the molar ratio of 1.5:3.5:95 (Eu3+/Tb3+/Gd3+) and the corresponding CIE coordinate (0.334, 0.336), which is very close to the coordinate for pure white-light (0.333, 0.333), thus ideal for white-light emission according to the 1931 CIE coordinate diagram (Fig. 6 and S16†).
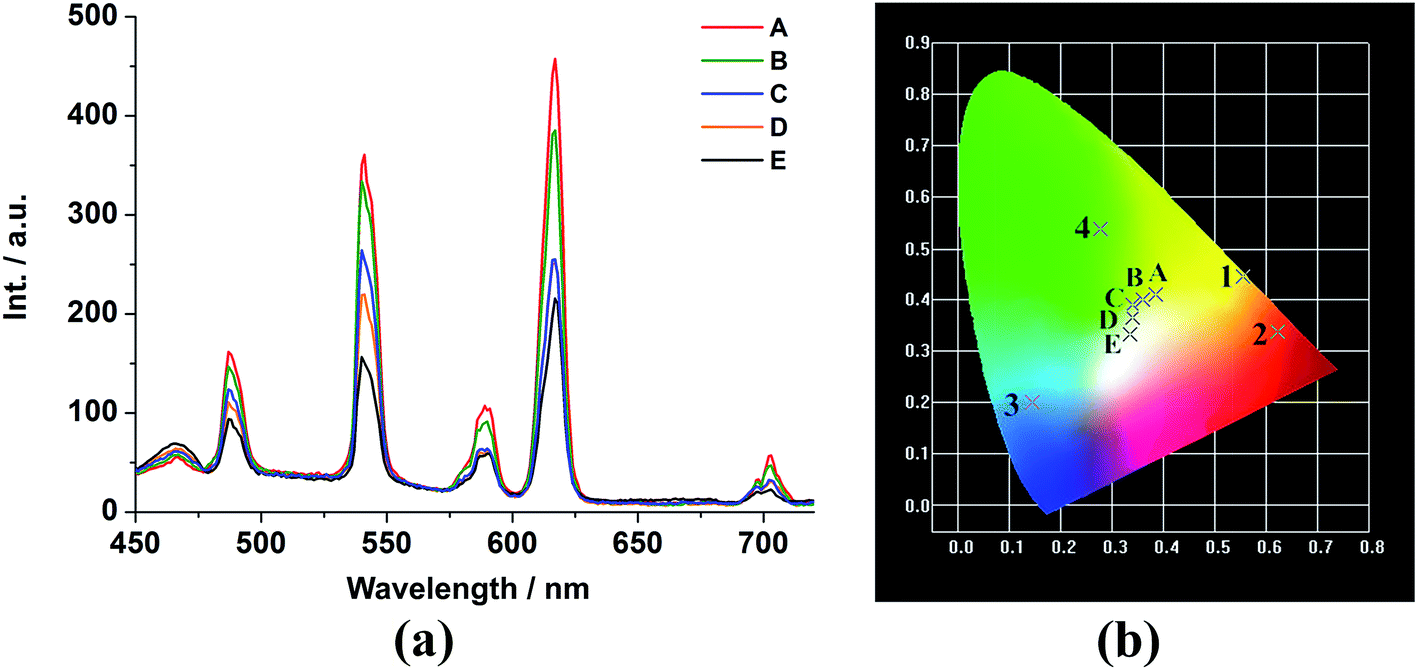 | ||
| Fig. 6 (a) Emission spectra and (b) CIE chromaticity diagram for 1, 2, 3, 4 and the doped EuxTbyGd1−x−y–BBTC (A–E) complexes excited at 340 nm. A–E (Eu, Tb, Gd%): A (4.5, 5.5, 90), B (4, 5, 91), C (3, 4.5, 92.5), D (2.5, 4, 93.5) and E (1.5, 3.5, 95). | ||
Moreover, it should be noted that the luminescent emission intensity of Eu3+ at 615 nm (corresponding to the transition 5D0 → 7F2) is 2.3-fold lower in the co-doped Eu0.015Tb0.035Gd0.95–BBTC complex than that in the pure complex Eu–BBTC (2), which is mainly due to the lower concentration of the Eu3+ ions (Fig. S17†). However, the emission intensity of Tb3+ at 542 nm (corresponding to the transition 5D4 → 7F5) decreases significantly and displays a 6.4-fold lower intensity compared to that in complex Tb–BBTC (4) (Fig. S17†), suggesting there is energy transfer from Tb3+ to Eu3+. Furthermore, the luminescent lifetimes of Eu3+ and Tb3+ emissions in this co-doped complex are 1.42 ms and 0.20 ms, respectively (Fig. S18†). Compared to the pure complex Eu–BBTC (2) and Tb–BBTC (4), the lifetime of Eu3+ in the co-doped complex is longer, while that of Tb3+ is shorter, further demonstrating that there exists intermetallic energy transfer from Tb3+ to Eu3+ centers in the co-doped complex, which is in agreement with others' reports.37
Conclusion
In summary, seven isostructural lanthanide–MOFs, [Ln2(BBTC)1.5(DMF)4]·2DMF·4H2O [Ln = Sm (1), Eu (2), Gd (3), Tb (4), Dy (5), Er (6) and Yb (7)], based on the conjugated tetracarboxylate ligands of BBTC4−, were successfully synthesized under similar solvothermal conditions. Each lanthanide–MOFs exhibits a 3-D microporous network containing two different subunits of Ln2(CO2)4 and Ln2(CO2)2 connected by BBTC4−. Moreover, the Ln–MOFs (Ln = Sm, Eu, Tb, Dy, Er and Yb) displayed intense luminescence in solid state at room temperature and the emissions exhibit the typical f–f transitions of Sm3+, Eu3+, Tb3+, Dy3+, Er3+ and Yb3+ ions, respectively, further indicating that there exist efficient energy transfer from the conjugated tetracarboxylate ligands to the Ln3+ ions and the conjugated BBTC4− ligand is a good “antenna effect” ligand for constructing luminescent lanthanide–MOFs. Compound 2, 3, and 4 acting as red-, blue-, and green-light emitting components are incorporated into the resulting tunable and white-light emitting materials by adjusting the relative concentration of Eu3+ and Tb3+ ions in the Gd3+ complex, suggesting that the identical coordination behavior of the different lanthanide ions allows in situ doping of various lanthanide ions into a parent MOFs, and the color of luminescence of the isostructural materials can be fine-tuned. This work provided the bright promise to exploit functional Ln–MOFs materials. It is expected that some more extensive endeavors to explore and develop the applications of solid state white-light emitting Ln–MOFs materials in lighting, imaging, agriculture, communication, and medicine will be carried out in the near future.Acknowledgements
Authors thank National Nature Science Foundation of China for financial support (grant no. 21371091 and 21371150).References
- (a) S. V. Eliseeva and J. C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189 RSC; (b) Y. Cui, Y. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed; (c) H. N. Li, H. Y. Li, L. K. Li, L. Xu, K. Hou and S. Q. Zang, Cryst. Growth Des., 2015, 15, 4331 CrossRef CAS.
- (a) M. Halim, M. S. Tremblay, S. Jockusch, N. J. Turro and D. Sames, J. Am. Chem. Soc., 2007, 129, 7704 CrossRef CAS PubMed; (b) I. Aillaud, J. Collin, C. Duhayon, R. Guillot, D. Lyubov, E. Schulz and A. Trifonov, Chem.–Eur. J., 2008, 14, 2189 CrossRef CAS PubMed; (c) L. L. Shen, L. Yang, Y. Fan and L. Wang, CrystEngComm, 2015, 17, 9363 RSC.
- (a) B. L. Chen, L. B. Wang, Y. Q. Xiao, F. R. Fronczek, M. Xue, Y. J. Cui and G. D. Qian, Angew. Chem., Int. Ed., 2009, 48, 500 CrossRef CAS PubMed; (b) A. Thibon and V. C. Pierre, J. Am. Chem. Soc., 2009, 131, 434 CrossRef CAS PubMed; (c) G. T. Wang, J. C. Zhang, Z. Y. Tang, H. T. Zhou, P. Zou and G. F. Hou, CrystEngComm, 2016, 18, 2437 RSC.
- (a) K. Hanaoka, K. Kikuchi, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2004, 126, 12470 CrossRef CAS PubMed; (b) B. L. Chen, Y. Yang, F. Zapata, G. Z. Lin, G. D. Qian and E. M. Lobkovsky, Adv. Mater., 2007, 19, 1693 CrossRef CAS; (c) J. Cepeda, S. Pérez-Yáñez, G. Beobide, O. Castillo, J. Á. García and A. Luque, Eur. J. Inorg. Chem., 2015, 4318 CrossRef CAS.
- (a) L. Zhao, S. Y. Qu, C. He, R. Zhang and C. Y. Duan, Chem. Commun., 2011, 47, 9387 RSC; (b) Y. Liu, X. Wu, C. He, Y. Jiao and C. Y. Duan, Chem. Commun., 2009, 48, 7554 RSC; (c) X. Zhu, C. He, D. Dong, Y. Liu and C. Y. Duan, Dalton Trans., 2010, 39, 10051 RSC; (d) W. Huang, D. Y. Wu, D. Guo, X. Zhu, C. He, Q. J. Meng and C. Y. Duan, Dalton Trans., 2009, 2081 RSC; (e) G. J. He, D. Guo, C. He, X. L. Zhang, X. W. Zhao and C. Y. Duan, Angew. Chem., Int. Ed., 2009, 48, 6132 CrossRef CAS PubMed; (f) X. H. Zhou, Y. H. Peng, X. D. Du, C. F. Wang, J. L. Zuo and X. Z. You, Cryst. Growth Des., 2009, 9, 1028 CrossRef; (g) J. J. Li, T. T. Fan, X. L. Qu, H. L. Han and X. Li, Dalton Trans., 2016, 45, 2924 RSC.
- (a) X. L. Zheng, Y. Liu, M. Pan, X. Q. Lü, J. Y. Zhang, C. Y. Zhao, Y. X. Tong and C. Y. Su, Angew. Chem., Int. Ed., 2007, 46, 7399 CrossRef CAS PubMed; (b) M. Pan, X.-L. Zheng, Y. Liu, W.-S. Liu and C.-Y. Su, Dalton Trans., 2009, 2157 RSC; (c) R. Huo, X. Li and D. Ma, Eur. J. Inorg. Chem., 2015, 852 CrossRef CAS.
- (a) J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220 CrossRef CAS PubMed; (b) S. Freslon, Y. Luo, C. Daiguebonne, G. Calvez and K. Bernot, Inorg. Chem., 2016, 55, 794 CrossRef CAS PubMed.
- (a) A. de Bettencourt-Dias, P. S. Barber and S. Bauer, J. Am. Chem. Soc., 2012, 134, 6987 CrossRef CAS PubMed; (b) S. Y. Wang, W. M. Wang, H. X. Zhang, H. Y. Shen, L. Jiang, J. Z. Cui and H. L. Gao, Dalton Trans., 2016, 45, 3362 RSC.
- (a) C. M. Wang, Y. Y. Wu, Y. W. Chang and K. H. Li, Chem. Mater., 2008, 20, 2857 CrossRef CAS; (b) G. Zeng, S. H. Xing, X. R. Wang, Y. L. Yang, D. X. Ma, H. W. Liang and G. H. Li, Inorg. Chem., 2016, 55, 1089 CrossRef CAS PubMed.
- (a) Y. B. Dong, P. Wang, J. P. Ma, X. X. Zhao, H. Y. Wang, B. Tang and R. Q. Huang, J. Am. Chem. Soc., 2007, 129, 4872 CrossRef CAS PubMed; (b) W.-G. Lu, L. Jiang and T.-B. Lu, Cryst. Growth Des., 2010, 10, 4310 CrossRef CAS.
- (a) Z. Guo, X. Song, H. Lei, H. Wang, H. Xu, G. Qian, H. Zhang and B. Chen, Chem. Commun., 2015, 51, 376 RSC; (b) X. Song, S. Song, S. Zhao, Z. Hao, M. Zhu, X. Meng, L. Wu and H. Zhang, Adv. Funct. Mater., 2014, 24, 4034 CrossRef CAS.
- (a) J. He, M. Zeller, A. D. Hunter and Z. T. Xu, J. Am. Chem. Soc., 2012, 134, 1553 CrossRef CAS PubMed; (b) K. Liu, H. P. You, Y. H. Zheng, G. Jia, Y. J. Huang, M. Yang, Y. H. Song, L. H. Zhang and H. J. Zhang, Cryst. Growth Des., 2010, 10, 16 CrossRef CAS.
- (a) S.-M. Li, X.-J. Zheng, D.-Q. Yuan, A. Ablet and L.-P. Jin, Inorg. Chem., 2012, 51, 1201 CrossRef CAS PubMed; (b) N. Guo, Y. H. Zheng, Y. C. Jia, H. Qiao and H. P. You, J. Phys. Chem. C, 2012, 116, 1329 CrossRef CAS.
- X. T. Rao, Q. Huang, X. L. Yang, Y. J. Cui, Y. Yang, C. D. Wu, B. L. Chen and G. D. Qian, J. Mater. Chem., 2012, 22, 3210 RSC.
- (a) Q. Tang, S. Liu, Y. Liu, D. He, J. Miao, X. Wang, Y. Ji and Z. Zheng, Inorg. Chem., 2014, 53, 289 CrossRef CAS PubMed; (b) X. Ma, X. Li, Y. E. Cha and L. P. Jin, Cryst. Growth Des., 2012, 12, 5227 CrossRef CAS; (c) M. L. Ma, C. Ji and S. Q. Zang, Dalton Trans., 2013, 42, 10579 RSC.
- Y. Liu, M. Pan, Q.-Y. Yang, L. Fu, K. Li, S.-C. Wei and C.-Y. Su, Chem. Mater., 2012, 24, 1954 CrossRef CAS.
- (a) Y. X. Hu, S. C. Xiang, W. W. Zhang, Z. X. Zhang, L. Wang, J. F. Bai and B. L. Chen, Chem. Commun., 2009, 48, 7551 RSC; (b) D. Sun, S. Ma, J. M. Simmons, J. R. Li, D. Yuan and H. C. Zhou, Chem. Commun., 2010, 46, 1329 RSC; (c) B. Zheng, Z. Liang, G. Li, Q. Huo and Y. Liu, Cryst. Growth Des., 2010, 10, 3405 CrossRef CAS; (d) D. Zhao, D. Yuan, A. Yakovenko and H. C. Zhou, Chem. Commun., 2010, 46, 4196 RSC; (e) B. Zheng, J. H. Luo, F. Wang, Y. Peng, G. H. Li, Q. S. Huo and Y. L. Liu, Cryst. Growth Des., 2013, 13, 1033 CrossRef CAS; (f) Z. Q. Liang, J. J. Du, L. B. Sun, J. Xu, Y. Mu, Y. Li, J. H. Yu and R. R. Xu, Inorg. Chem., 2013, 52, 10720 CrossRef CAS PubMed; (g) L. B. Sun, H. Z. Xing, J. Xu, Z. Q. Liang, J. H. Yu and R. R. Xu, Dalton Trans., 2013, 42, 5508 RSC.
- (a) L. F. Wang, L. C. Kang, W. W. Zhang, F. M. Wang, X. M. Ren and Q. J. Meng, Dalton Trans., 2011, 40, 9490 RSC; (b) L. Zhai, W. W. Zhang and X. M. Ren, Dalton Trans., 2015, 44, 5746 RSC; (c) J. Zhang, B. Zheng, T. T. Zhao, G. H. Li, Q. S. Huo and Y. L. Liu, Cryst. Growth Des., 2014, 14, 2394 CrossRef CAS.
- Software packages SMART and SAINT, Siemens Analytical X-ray Instrument Inc., Madison, WI, 1996 Search PubMed.
- G. M. Sheldrick, SHELX−97, Program for the refinement of crystal structure, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- (a) H. S. Wang, B. Zhao, B. Zhai, W. Shi, P. Cheng, D. Z. Liao and S. P. Yan, Cryst. Growth Des., 2007, 7, 1851 CrossRef CAS; (b) J. Xu, J. Cheng, W. Su and M. Hong, Cryst. Growth Des., 2011, 11, 2294 CrossRef CAS.
- (a) M. D. Allendorf, C. A. Bauer, R. K. Bhaktaa and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC; (b) D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502 CrossRef CAS PubMed.
- H.-B. Zhang, X.-C. Shan, Z.-J. Ma, L.-J. Zhou, M.-J. Zhang, P. Lin, S.-M. Hu, E. Ma, R.-F. Li and S.-W. Du, J. Mater. Chem. C, 2014, 2, 1367 RSC.
- A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127 CrossRef CAS.
- S. Su, W. Chen, C. Qin, S. Song, Z. Guo, G. Li, X. Song, M. Zhu, S. Wang, Z. Hao and H. Zhang, Cryst. Growth Des., 2012, 12, 1808 CAS.
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824 CrossRef.
- Phosphor Handbook, ed. W. M. Yen, S. Shionoya and H. Yamamoto, CRC Press, Boca Raton, 2007 Search PubMed.
- (a) K. Driesen, R. Van Deun, C. Görller-Walrand and K. Binnemans, Chem. Mater., 2004, 16, 1531 CrossRef CAS; (b) K. Lunstroot, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Görller-Walrand, K. Binnemans and K. Driesen, Inorg. Chem., 2009, 48, 3018 CrossRef CAS PubMed.
- (a) J. Feng, H. J. Zhang, S. Y. Song, Z. F. Li, L. N. Sun, Y. Xing and X. M. Guo, J. Lumin., 2008, 128, 1957 CrossRef CAS; (b) J. W. Stouwdam and F. C. J. M. van Veggel, Nano Lett., 2002, 2, 733 CrossRef CAS.
- P. Martín-Ramos and C. Coya, J. Phys. Chem. C, 2013, 117, 10020 Search PubMed.
- (a) L. N. Sun, W. Mai and S. Dang, J. Mater. Chem., 2012, 22, 5121 RSC; (b) L. N. Sun, Y. N. Qiu, T. Liu, J. Feng, W. Deng and L. Y. Shi, Luminescence, 2015, 30, 1071 CrossRef CAS PubMed.
- S. Comby and J.-C. G. Bünzli, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, Amsterdam, 2007, vol. 37, ch. 235, p. 217 Search PubMed.
- A. Rodríguez-Diéguez, A. Salinas-Castillo, A. Sironi, J. M. Seco and E. Colacio, CrystEngComm, 2010, 12, 1876 RSC.
- (a) W. H. Di, X. J. Wang, P. F. Zhu and B. J. Chen, J. Solid State Chem., 2007, 180, 467 CrossRef CAS; (b) L. Zhai, W. W. Zhang, X. M. Ren and J. L. Zuo, J. Solid State Chem., 2015, 230, 14 CrossRef CAS.
- J. W. Rong, W. W. Zhang and J. F. Bai, CrystEngComm, 2016, 18, 7728 RSC.
- (a) D. Ma, X. Li and R. Huo, J. Mater. Chem. C, 2014, 2, 9073 RSC; (b) Y. Luo, Y. J. Zheng, G. Calvez, S. Freslon, K. Bernot, C. Daiguebonne, T. Roisnel and O. Guillou, CrystEngComm, 2013, 15, 706 RSC; (c) C. Gao, A. M. Kirillov, W. Dou, X. Tang, L. Liu, X. Yan, Y. Xie, P. Zang, W. Liu and Y. Tang, Inorg. Chem., 2014, 53, 935 CrossRef CAS PubMed; (d) P. C. R. Soares-Santos, L. Cunha-Silva, F. A. A. Paz, R. A. S. Ferreira, J. Rocha, T. Trindade, L. D. Carlos and H. I. S. Nogueira, Cryst. Growth Des., 2008, 8, 2505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The preparation of 2–7 is detailed in the supporting material. Elemental analysis and main IR bands for 1–7 are given in Table S1. PXRD patterns of Gd–BBTC, Eu0.25Gd0.75–BBTC and Tb0.5Gd0.5–BBTC are given in Fig. S1. PXRD patterns for complex 3 and co-doped compounds EuxTbyGd1−x−y–BBTC are shown in Fig. S2. Elemental analysis for EuxGd1−x–BBTC and TbyGd1−y–BBTC monodoped compounds are shown in Table S2. Elemental analysis for EuxTbyGd1−x−y–BBTC co-doped compounds are presented in Table S3. EDX-spectra of Eu0.25Gd0.75–BBTC, Tb0.5Gd0.5–BBTC and Eu0.045Tb0.055Gd0.9–BBTC are given in Fig. S3. ICP analysis for EuxGd1−x–BBTC, TbyGd1−y–BBTC and EuxTbyGd1−x−y–BBTC co-doped compounds are shown in Table S4. The main data of collection and refinement details of complexes 1 and 3 are given in Table S5. Selected bond lengths and angles for 1–7 are given in Tables S6 and S7. PXRD patterns of 1–7 are plotted in Fig. S4. The IR spectra of 1–7 are presented in Fig. S5. The crystal structures of 1, 2, 3, 5, 6 and 7 are shown in Fig. S6. Space filled representation of the 3-D framework in 4 is shown in Fig. S7. The TG curves of 1–7 are presented in Fig. S8. The UV-vis absorption and emission spectra of ligand H4BBTC are shown in Fig. S9. The excitation spectra of 2, 3, 4 and 5 are shown in Fig. S10. Emission spectra of 1 and 5, and NIR emission spectra of 5, 6 and 7 are shown in Fig. S11. Luminescence decay profiles for 1, 5, 6 and 7 are presented in Fig. S12. Emission spectra and CIE chromaticity diagram for EuxGd1−x–BBTC are shown in Fig. S13. CIE coordinates of the EuxGd1−x–BBTC are given in Table S8. Emission spectra and CIE chromaticity diagram for TbyGd1−y–BBTC are given in Fig. S14. CIE coordinates of the TbyGd1−y–BBTC are presented in Table S9. The excitation spectra of Eu0.25Gd0.75–BBTC, Tb0.5Gd0.5–BBTC, and Eu0.015Tb0.035Gd0.95–BBTC are shown in Fig. S15. Emission spectra and CIE chromaticity diagram for Eu0.015Tb0.035Gd0.95–BBTC are given in Fig. S16. CIE coordinates of the EuxTbyGd1−x−y–BBTC are presented in Table S10. Emission spectra of complexes 2, 4 and the doped Eu0.015Tb0.035Gd0.95–BBTC complex are shown in Fig. S17. Luminescence decay profiles of Eu3+ and Tb3+ in the Eu0.015Tb0.035Gd0.95–BBTC doped complex are presented in Fig. S18. CCDC 1478788, 1478790, 1478791, 1478792 and 1478793 are for 2, 4, 5, 6 and 7, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra22001k |
| This journal is © The Royal Society of Chemistry 2016 |