
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetics and mechanism of the OH-radical and Cl-atom oxidation of propylene carbonate†
Ian
Barnes
*,
Peter
Wiesen
and
Michael
Gallus
University of Wuppertal, School of Mathematics and Natural Sciences, Institute for Atmospheric and Environmental Research, Gauss Strasse 20, 42119 Wuppertal, Germany. E-mail: barnes@uni-wuppertal.de; Fax: +49 202 4392505; Tel: +49 202 439 2510
First published on 10th October 2016
Abstract
Rate coefficients have been measured at 298 K and atmospheric pressure for the reaction of OH radicals and Cl atoms with propylene carbonate. The measurements were performed in a large volume photoreactor using in situ FTIR spectroscopy for the analysis. The following rate coefficients (in units of cm3 per molecule per s) were obtained: k(OH + PC) = (2.52 ± 0.51) × 10−12 and k(Cl + PC) = (1.77 ± 0.43) × 10−11. Product studies performed on the OH-radical and Cl-atom mediated oxidation of propylene carbonate in air support that the major fate of the intermediate cyclo-methyl-pentoxy radicals, formed in the degradation reaction sequence, is unimolecular decomposition. The FTIR product spectra, in combination with the absence of other potential products, suggest that the decomposition probably results to a large extent in the formation of acetyl formyl carbonate, CH3C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OC(O)OCH(O). In product studies performed in N2, in which ppm levels of O2 are present, formation of acetic acid was observed in addition to acetyl formyl carbonate. It is postulated that the acid formation occurs via a pathway involving a 1,3-hydrogen shift mechanism with an intermediate alkoxy radical which is able to compete with the unimolecular decomposition pathway of the alkoxy radical at very low O2 partial pressures.
O)OC(O)OCH(O). In product studies performed in N2, in which ppm levels of O2 are present, formation of acetic acid was observed in addition to acetyl formyl carbonate. It is postulated that the acid formation occurs via a pathway involving a 1,3-hydrogen shift mechanism with an intermediate alkoxy radical which is able to compete with the unimolecular decomposition pathway of the alkoxy radical at very low O2 partial pressures.
Introduction
Propylene carbonate (PC) is a carbonate ester, derived from propylene glycol, with the empirical formula CH3C2H3O2CO and molecular structure: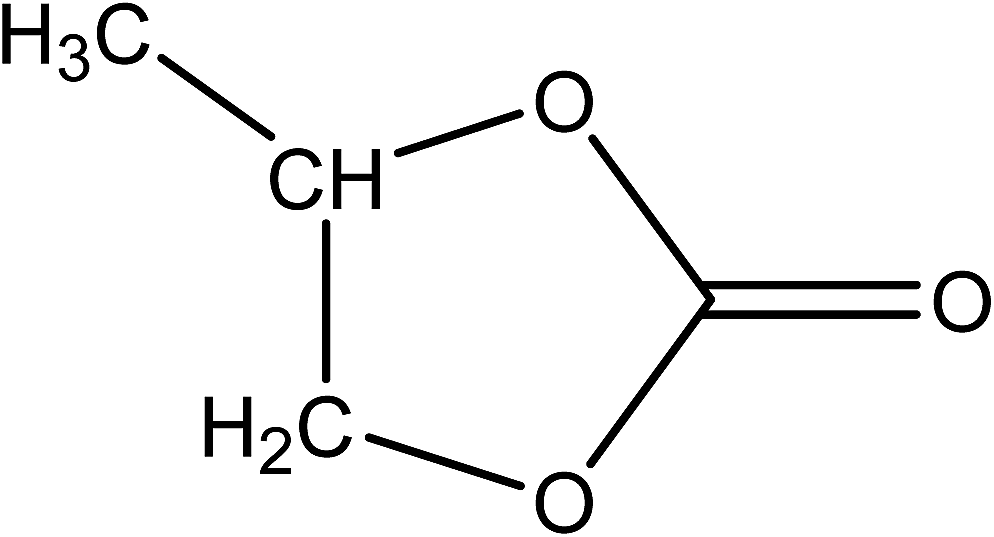
It is a colourless to yellowish and odourless liquid with a high boiling point. It is used as a polar, aprotic solvent in many applications and is present in adhesives, cosmetics and personal care products, for example, it is used in the formulation of makeup, mainly lipstick, eye shadow, and mascara.1–3 It is chiral but is used exclusively as the racemic mixture.
The production of propylene carbonate and its widespread use as a solvent and chemical intermediate will result in fugitive releases to the environment. Propylene carbonate has a vapour pressure of 0.045 mm Hg at 25 °C (ref. 4) and based on this assessments of its possible environmental fate have concluded that if released to the atmosphere it will exist solely as a vapour.5
As for the majority of volatile organic compounds (VOCs) in the atmosphere vapour-phase propylene carbonate will be degraded in the atmosphere to a large extend by reaction with hydroxyl radicals.6 Direct loss of propylene carbonate by photolysis is also potentially possible since it contains a functional group that can absorb light greater than 290 nm, however, nothing is currently known on the atmospheric photolysis frequency of propylene carbonate which would allow an evaluation of the importance of this loss process compared to reaction with OH. Using a structure–activity relationship7 a rate coefficient of 3.78 × 10−12 cm3 per molecule per s has been estimated for the reaction of OH radicals with propylene carbonate, which corresponds to an atmospheric lifetime for the compound of between 3 to 4 days. To the best of our knowledge there have been to date no experimental determinations of the OH rate coefficient for the reaction.
Conventionally, it has been thought that Cl-atom mediated atmospheric oxidations of VOCs was only important in coastal and marine regions,8–10 however, in recent years significant concentrations of Cl-atom precursors such as nitryl chloride (ClNO2) have been measured in continental regions far removed from coastal regions.11–15 These findings suggest that the Cl-atom mediated VOC oxidation chemistry may be much more prevalent than previously thought. Since a rate coefficient for the reaction of Cl-atoms with propylene carbonate does not exist in the literature and the reaction may possibly have some atmospheric significant it has been investigated in this work.
In summary, the objectives of the present work have been to investigate for the first time the kinetics and mechanism of the OH-radical and Cl-atom mediated photooxidation of propylene carbonate and assess any possible environmental consequences. Apart from any atmospheric relevance of the results from the present work it also provides mechanistic insight into the gas-phase fate of the cyclo-methyl-pentoxy radicals that are formed in the OH- and Cl-initiated photooxidation of propylene carbonate.
Experimental
The experiments were performed in a 1080 L quartz-glass photoreactor in synthetic air at a total pressure of 760 Torr (760 Torr = 101.325 kPa). Since the photoreactor is described in detail elsewhere16 only a brief general overall description is given here. A pumping system consisting of a turbo-molecular pump backed by a double stage rotary fore pump allows the photoreactor to be evacuated to 10−3 Torr. Magnetically coupled Teflon mixing fans are mounted inside the chamber to ensure homogeneous mixing of the reactants. Two types of lamps are available for photolysis of the radical/atom precursors: 32 super actinic fluorescent lamps (Philips TL 05/40 W: 320 < λ < 480 nm, λmax = 360 nm) and 32 low-pressure mercury lamps (Philips TUV/40 W, λmax = 254 nm). The lamps are distributed evenly around the photoreactor, are wired in parallel, and can be switched individually. A white type multiple-reflection mirror system with a total optical path length of 484.7 ± 0.8 m is mounted inside the photoreactor for sensitive in situ long path absorption monitoring of reactants and products in the IR spectral range 4000–700 cm−1. IR spectra were recorded with a spectral resolution of 1 cm−1 using a Nicolet Nexus FT-IR spectrometer equipped with a KBr beam splitter and a liquid nitrogen cooled mercury–cadmium–telluride (MCT) detector.Rate coefficients for the reactions of OH radicals and Cl atoms with propylene carbonate were determined using the relative kinetic technique. Hydroxyl radicals were produced by the photolysis of hydrogen peroxide using the mercury lamps:
| H2O2 + hν (λ = 254 nm) → 2OH | (1) |
Chlorine atoms were generated by photolysis of molecular Cl2 with the fluorescent lamps:
| Cl2 + hν (320 < λ < 480 nm) → 2Cl | (2) |
In the presence of OH radicals or Cl atoms, propylene carbonate and the reference compounds decay through the following reactions:
| OH/Cl + PC → products, kPC | (3) |
| OH/Cl + reference → products, kref | (4) |
Provided that the reference compounds and the propylene carbonate are lost only by reactions (3) and (4), then it can be shown that:
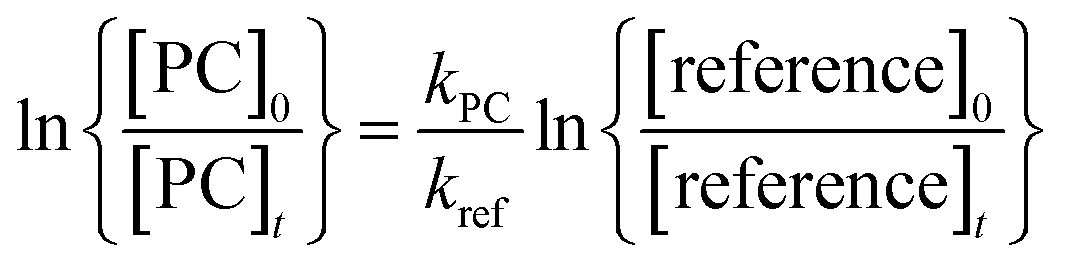 | (I) |
The relative rate technique relies on the assumption that both propylene carbonate and the reference compounds are removed solely by reaction with either OH radicals or Cl atoms. In order to verify this assumption various tests were performed. Mixtures of propylene carbonate and the reference compounds with either H2O2 or molecular chlorine were prepared and allowed to stand in the dark for 30 minutes the duration of a typical experiment. Neither reaction of the radical precursors (H2O2/Cl2) with propylene carbonate nor any of the reference compounds was observed. Wall loss of all the substances was also insignificant. To test for possible photolysis loss of propylene carbonate it was irradiated alternatively in air with the fluorescent and mercury lamps. Neither type of lamp caused photolytic loss of propylene carbonate.
The initial concentrations of propylene carbonate and the reference compounds methanol, n-butane and ethene were 2–4 ppmV (1 ppmV = 2.46 × 1013 molecule per cm3 at 298 K and 760 Torr of total pressure). The initial concentrations of H2O2 and Cl2 were typically around 10 and 5 ppmV, respectively. The experiments were performed in 760 Torr of synthetic air at (298 ± 2) K. In a typical experiment 60 interferograms were co-added per spectrum over a period of approximately 1 minute and 15–20 such spectra were recorded per experiment. The first 5 spectra were always recorded without lamps to check that wall loss of the reactants remained negligible.
Reactants and products were quantified by comparison with calibrated reference spectra contained in the IR spectral databases of the laboratory in Wuppertal. Quantitative spectral subtraction was accomplished using the spectral subtraction option in the OMNIC Software Suite 8.0 from Thermo Scientific. The reactants were monitored at the following infrared absorption frequencies (in cm−1): propylene carbonate 1866, n-butane 2965, methanol 1033 and ethene 950. The reaction products were monitored at the following infrared spectra (in cm−1): formaldehyde 2766, acetic acid 3581 and 1798, formic acid 1776 and 1105 and carbon monoxide 2162.
The chemicals used in the experiments had the following purities as given by the manufacturer and were used as supplied: synthetic air (Air Liquide, 99.999%), Cl2 (Messer Griesheim, 2.8), n-butane (Messer Griesheim, 2.5), ethene (Messer Schweiz AG, 3.5), methanol (Sigma-Aldrich, ≥99.8%) hydrogen peroxide (Interox, 85%), propylene carbonate (Sigma-Aldrich, 98%).
Results and discussion
Kinetic study
Fig. 1(A) shows exemplary plots of the kinetic data obtained for the reaction of OH radicals with propylene carbonate measured relative to n-butane and ethene. Fig. 1(B) shows an exemplary plot of the kinetic data obtained for the reaction of Cl atoms with propylene carbonate measured relative to methanol and ethene. Reasonable straight lines were obtained for both reactions using the two reference compounds. The rate coefficient ratios kPC/kref obtained from linear regression analyses of the plots of the kinetic data are given in Table 1 for the reaction of OH and Cl with propylene carbonate. The ratios are the averages from at least three experiments with each reference compound and the errors are the 2σ standard deviation. The rate coefficients kPC given in Table 1 were put on an absolute basis using the following values for the reactions of the reference compounds (in units of cm3 per molecule per s): k(OH + n-butane) = 2.36 × 10−12;17k(OH + ethene) = 7.9 × 10−12;18k(Cl + CH3OH) = 5.5 × 10−11;18k(Cl + ethene) = 1.1 × 10−10.18 | ||
| Fig. 1 Plot of the kinetic data for the reaction of (A) OH with propylene carbonate measured relative to n-butane and ethene and (B) Cl with propylene carbonate measured relative to methanol and ethene. | ||
| Reaction | Reference | k PC/kref | k PC (cm3 per molecule per s) | SAR |
|---|---|---|---|---|
| a Calculated using the OH SAR of Kwok and Atkinson.19 b Estimated using the Cl SAR of Aschmann and Atkinson22 and a substituent factor of F(RC(O)O–) = 0.66 for esters reported by Xing et al.23 (see text). | ||||
| OH + PC | n-Butane | 0.987 ± 0.089 | (2.33 ± 0.21) × 10−12 | 3.78 × 10−12a |
| 1.027 ± 0.103 | (2.42 ± 0.24) × 10−12 | |||
| 0.991 ± 0.079 | (2.34 ± 0.19) × 10−12 | |||
| 1.029 ± 0.113 | (2.43 ± 0.27) × 10−12 | |||
| Average | (2.38 ± 0.30) × 10 −12 | |||
| Ethene | 0.316 ± 0.032 | (2.50 ± 0.25) × 10−12 | ||
| 0.355 ± 0.041 | (2.80 ± 0.32) × 10−12 | |||
| 0.341 ± 0.031 | (2.69 ± 0.25) × 10−12 | |||
| Average | (2.66 ± 0.44) × 10 −12 | |||
| Cl + PC | Methanol | 0.315 ± 0.025 | (1.73 ± 0.14) × 10−11 | 1.64 × 10−11b |
| 0.370 ± 0.038 | (2.04 ± 0.21) × 10−11 | |||
| 0.304 ± 0.033 | (1.67 ± 0.18) × 10−11 | |||
| 0.318 ± 0.038 | (1.75 ± 0.21) × 10−11 | |||
| Average | (1.81 ± 0.38) × 10 −11 | |||
| Ethene | 0.141 ± 0.014 | (1.55 ± 0.15) × 10−11 | ||
| 0.150 ± 0.017 | (1.65 ± 0.19) × 10−11 | |||
| 0.179 ± 0.018 | (1.97 ± 0.20) × 10−11 | |||
| Average | (1.72 ± 0.39) × 10 −11 | |||
Since the values of the rate coefficients obtained for the reaction of OH with propylene carbonate using n-butane and ethene as reference compounds are in relatively good agreement we prefer to quote a final rate coefficient for the reaction of (2.52 ± 0.51) × 10−12 cm3 per molecule per s which is the average of all the individual determinations. Similarly for the reaction of Cl with propylene carbonate because of the good agreement between the determinations with the two reference compounds we prefer to quote a final rate coefficient for the reaction of (1.77 ± 0.43) × 10−11 cm3 per molecule per s which is the average of all the determinations.
There are no other kinetic studies in the literature with which the measured rate coefficients for the reactions of OH radicals and Cl atoms with PC can be compared. The OH structure activity relationship (SAR) of Kwok and Atkinson19 predicts a value of 3.78 × 10−12 cm3 per molecule per s for the reaction of OH with PC which is ∼60% higher than the measured value. However, since the substituent factor F(–OC(O)R) was used in the calculation to represent the carbonate group (–O(CO)O–) and the ring-strain factor for a C5 ring was used, the agreement between experiment and estimate can be considered as reasonable. The rate coefficient for the reaction of OH with PC can be compared to that of OH with γ-valerolactone for which a value of (2.81 ± 0.34) × 10−12 cm3 per molecule per s has been reported,20i.e. approximately 16% higher than that of OH with PC. These compounds differ in that the O atom adjacent to the –CH2– entity in PC is a –CH2– entity in γ-valerolactone, therefore, one would expect a somewhat higher OH rate coefficient for γ-valerolactone compared to PC because of the presence of the extra –CH2– entity. Just how much higher the rate coefficient for the reaction of OH with γ-valerolactone should be is hard to gauge because of the uncertainty in factors for substituent effects and the ring strains for PC and γ-valerolactone. The OH SAR of Kwok and Atkinson19 predicts that both compounds should be equally reactive toward OH. This prediction serves to show that the rate coefficient determined for the reaction of OH with PC in this study is of the correct order of magnitude.
A similar comparison can also be made for the reactions of Cl with PC and γ-valerolactone. A rate coefficient of (3.74 ± 0.22) × 10−11 cm3 per molecule per s has been measured for the reaction of Cl with γ-valerolactone in our laboratory.21 In this case the reaction of Cl with γ-valerolactone is just over a factor of two higher than the value measured for Cl with PC in this study. In an attempt to estimate the rate coefficient for the reaction of Cl with PC we have used the approach adopted in the OH SAR of Kwok and Atkinson,19i.e. we have used a substituent factor F(RC(O)O–) as a surrogate for the carbonate –O–C(O)O– entity. Using the parameters given in the Cl SAR of Aschmann and Atkinson22 and the substituent factor F(RC(O)O–) = 0.066 reported by Xing et al.23 for the reaction of Cl with esters we estimate a value of 1.64 × 10−11 cm3 per molecule per s for the reaction of Cl with PC. This value is in surprisingly good agreement with the experimental value and supports that using the substituent factor F(RC(O)O–) as a surrogate for the carbonate –O–C(O)O– entity is justified.
Product study
The products formed in the Cl-atom initiated photooxidation of propylene carbonate have been investigated in one atmosphere of synthetic air and nitrogen. Fig. 2, trace (A) shows a spectrum of propylene carbonate and trace (B) shows the product spectrum obtained after reaction with Cl and subtraction of residual propylene carbonate. Traces (C)–(E) show reference spectra of HCl, CO and acetic anhydride. In the product spectrum the formation of HCl and CO in the spectral regions around 3000 and 2000 cm−1, respectively, are clearly visible. A product spectrum with the absorptions of HCl and CO removed is not shown since they do not interfere with the main product absorptions below 2000 cm−1 and product absorptions above 2000 cm−1 are negligible.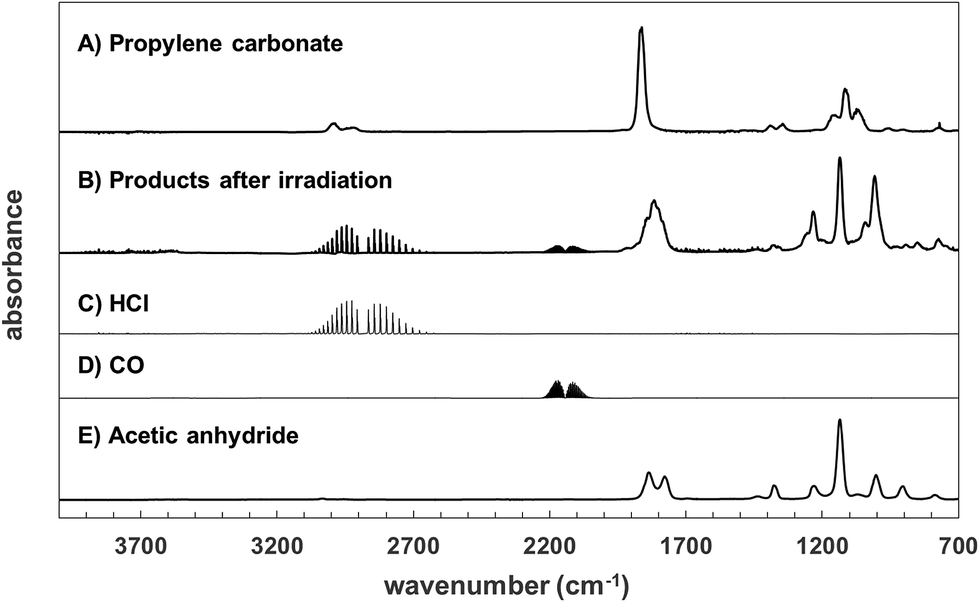 | ||
| Fig. 2 Products formed in the irradiation of a propylene carbonate/Cl2 mixture in synthetic air. Trace (A) is a spectrum of propylene carbonate, trace (B) is a product spectrum after irradiation and subtraction of excess propylene carbonate and traces (C)–(E) are reference spectra of HCl, CO and acetic anhydride. | ||
The product spectrum is relatively simple indicating the probable dominance of one major product. The spectrum contains a broad peak in the carbonyl region from 1900 to 1725 cm−1 with two apparent maxima at approximately 1815 and 1844 cm−1. The fingerprint region is dominated by 3 absorptions with maxima at 1232, 1314 and 1007 cm−1. Also present in the spectrum, but not visible in trace (B) are weak absorptions due to formic acid (HC(O)OH). The concentration–time profiles of propylene carbonate and the identified products HCl, CO and HC(O)OH are shown in Fig. 3. The errors on the product concentrations were typically ∼5%, for better clarity they have not been included in Fig. 3
 | ||
| Fig. 3 Concentration–time profile for the decay of propylene carbonate and the formation of products on irradiation of a propylene carbonate/Cl2/air mixture. | ||
Since the reaction of Cl with propylene carbonate proceeds by H-atom abstraction the formation of HCl is expected. Although the formation of HC(O)OH appears to be primary in nature we can not think of a plausible mechanism for a primary formation route and think it may stem from the rapid decomposition of an unstable primary product such as acetyl formyl carbonate (see below). The small amount of CO observed in the system is definitely being formed in secondary reactions. The strong product absorptions in the carbonyl and fingerprint regions all correlate linearly with the absorption of propylene carbonate over most of the reaction period, however, when most of the propylene carbonate has been consumed loss (probably wall) of the product(s) giving rise to the absorptions is evident. Fig. S1, panel A, in the ESI† compares the absorbance-time behaviour of the propylene carbonate carbonyl absorption at 1867 cm−1 with that of one of the product absorbance's at 1009 cm−1. In Fig. S1,† panel B, the absorbance of the propylene carbonate carbonyl absorption at 1867 cm−1 is plotted against product absorption at 1009 cm−1 and demonstrates the linear correlation over most of the reaction period.
Exactly similar results were obtained with OH as the oxidant, however, since (i) both OH and Cl react by similar mechanisms with propylene carbonate, i.e. H-atom abstraction,5 (ii) the conversions of propylene carbonate were much lower and (iii) the OH product spectra were difficult to analyse due to strong absorptions from H2O2 and water we are only presenting here the results with Cl as oxidant.
The OH SAR of Kwok and Atkinson19 predicts contributions of around 4, 31 and 65% for H-atom abstraction from the primary, secondary and tertiary hydrogens in propylene carbonate. It is not possible to estimate accurately the corresponding percentages for H-atom abstraction with Cl atoms since reliable substituent factors are not available to account for the effect of the cyclic –OC(O)O– functionality. However, the good agreement between the product spectra obtained using both Cl and OH and the similarity in reaction mechanism suggests that H-atom abstraction from the secondary and tertiary hydrogens will also dominate for the reaction of Cl with propylene carbonate. This borne out by the interpretation of the results discussed below.
The radicals formed from H-atom abstraction from the primary, secondary and tertiary hydrogens in propylene carbonate will add O2 to form the corresponding peroxy radicals. The main but not solitary fate of the peroxy radicals will be self and reaction with other peroxy radicals to form the corresponding alkoxy radicals,24,25 which in the cases of the radicals formed from secondary and tertiary H-atom abstraction, will be cyclo-methyl-pentoxy radicals. Reaction channels forming molecular products are also possible24,25 but as will be discussed below these are thought to be relatively minor for the cyclic peroxy radicals involved in the degradation of propylene carbonate. The alkoxy radicals that can be formed in the reaction of Cl/OH with propylene carbonate are shown in Fig. 4.
 | ||
| Fig. 4 Alkoxy radicals formed through H-atom abstraction by Cl atoms or OH radicals from the primary (a), secondary (b) and tertiary (c) hydrogens in propylene, carbonate. | ||
Scheme 1 outlines the possible reaction channels for the reactions of the alkoxy radical formed from H-atom abstraction at the methyl group in propylene carbonate. As depicted in Scheme 1 the radical could react with O2 to form an aldehydic carbonate and/or decompose to form a carbonate alkyl radical and HCHO. Further reactions of the alkyl radical could form a cyclic keto carbonate or glyoxal. If the carbonate group containing products were being formed to any appreciable extent a strong carbonyl absorption from this group should occur at around 1870 cm−1,26,27 for example, the carbonyl absorption from propylene carbonate occurs at 1866 cm−1 in the gas phase. However, in the product spectrum the carbonyl absorption is very weak in this region. Formation of HCHO and glyoxal was also not observed indicating that the decomposition pathways are negligible. Based on these observations we conclude that product formation from H-atom abstraction at the methyl group in propylene carbonate is very minor.
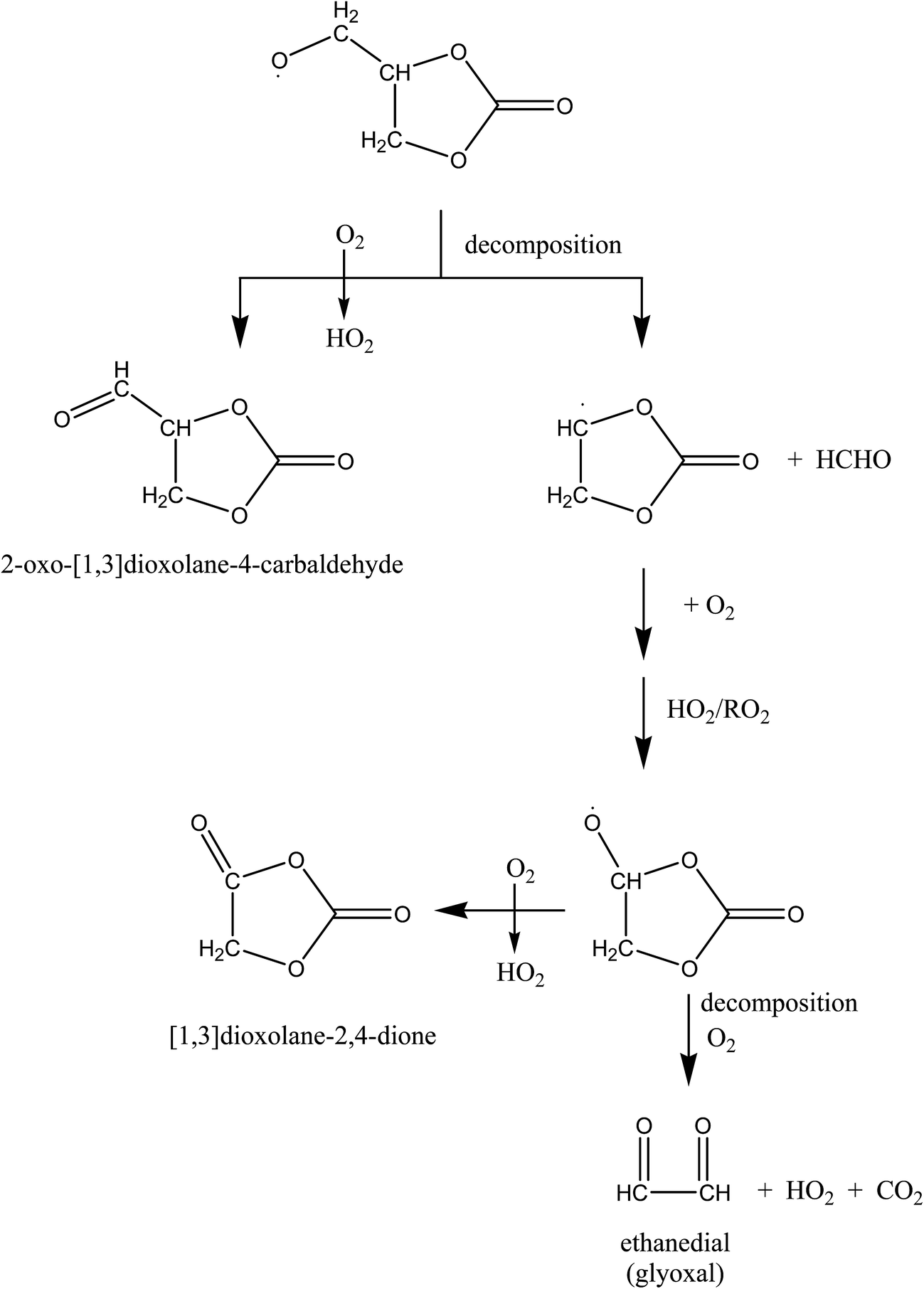 | ||
| Scheme 1 Possible reaction channels for the alkoxy radical formed after H-atom abstraction from the methyl group in propylene carbonate. | ||
Scheme 2 outlines possible reaction routes for the alkoxy radical formed from H-atom abstraction from the methylene group in propylene carbonate. The radical can react with O2 to form a keto carbonate compound or cleave the C–C bond in the ring to form the linear alkyl radical shown in Scheme 1. It is well established that the major fate of the cyclopentoxy radical is ring-opening rather than reaction with O2 (ref. 28–30) and it is expected that this is also case for the alkoxy radical formed at the methylene group in propylene carbonate. The absence of any strong carbonyl absorption at 1870 cm−1, as discussed above, also supports that formation of the molecular product through reaction of the radical with O2 is negligible.
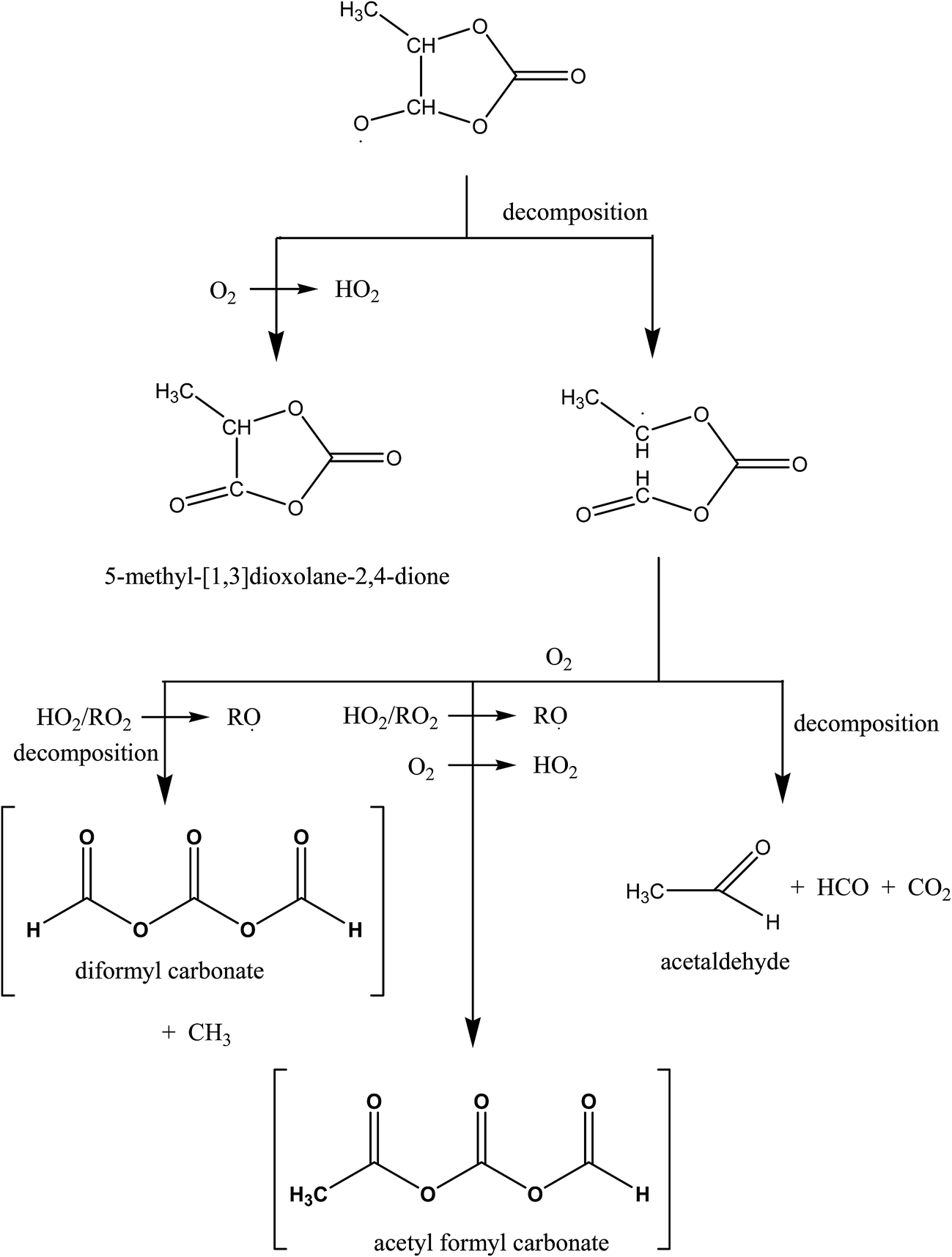 | ||
| Scheme 2 Possible reaction channels for the alkoxy radical formed after H-atom abstraction from the methylene group in propylene carbonate. Major suspected products are shown in brackets. | ||
The alkyl radical could decompose with formation of acetaldehyde or add O2 and through a sequence of peroxy–peroxy reactions etc. eventually form diformyl carbonate (CH3C(O)OC(O)OC(O)H) and acetic formyl carbonate (CH3C(O)OC(O)OC(O)CH3). Since formation of acetaldehyde is not observed and the further reactions of the CH3 radicals would result in the formation of HCHO and CH3OH, both of which were also not observed, it would appear that the major pathway must be formation of acetic formyl carbonate. The carbonyl absorption frequencies of open-chain carbonates occur at lower frequencies than those of the cyclic analogues.26,27 A shift to lower carbonyl frequencies compared to propylene carbonate is observed in the product spectrum obtained on reacting Cl with propylene carbonate (Fig. 2, trace (B)). The structure of acetic formyl carbonate contains an anhydride entity CH3–C(O)–O–C(O)– and this should be reflected in the product spectrum. The product spectrum is compared with a reference spectrum of acetic anhydride in Fig. 2, traces (B) and (E), respectively. It can be seen that the positions of the carbonyl absorptions and also those in the fingerprint region match very well. Acetic anhydride has two absorption maxima in the carbonyl region which are due to the symmetrical and asymmetrical stretching vibrations of the carbonyl groups. The carbonyl stretching region in the product spectrum from the reaction of Cl with propylene carbonate also shows the existence of different carbonyl stretching absorption maxima. The resolution in the carbonyl maxima, that is clearly evident in the infrared spectrum of acetic anhydride, is probably lost in the infrared spectrum of acetyl formyl carbonate because of the presence of the additional carbonyl functionality in acetic formyl carbonate.
Scheme 3 outlines possible reaction routes for the alkoxy radical formed from H-atom abstraction from the tertiary H-atom in propylene carbonate. The alkoxy radical could eject a methyl group and form a keto-cyclo-carbonate, however, the lack of a carbonate absorption in the product spectrum and also the presence of HCHO and CH3OH, which would be formed from further reactions of the methyl radical, supports that this reaction pathway is negligible. The major reaction pathway for this radical will be ring-opening for which there are two possibilities, i.e. either C–O or C–C bond cleavage. The C–O bond cleavage route would result in the formation of methyl glyoxal, however, as this is not observed in the product spectrum this pathway is considered to be negligible. The major pathway must then be C–C bond cleavage with formation once again of acetyl formyl anhydride.
 | ||
| Scheme 3 Possible reaction channels for the alkoxy radical formed after H-atom abstraction from the tertiary H-atom in propylene carbonate. Major suspected products are shown in brackets. | ||
In summary, the evidence from the product study supports that H-atom abstraction from both the secondary and tertiary hydrogens in propylene carbonate will lead predominately to the formation acetyl formyl carbonate.
A product study has been performed on the reaction of Cl with propylene carbonate in one atmosphere of nitrogen. It should be borne in mind, that although the reaction was performed in N2, in large volume photoreactors such as used in this work, ppm levels of O2 in the reaction system are unavoidable. The product spectrum obtained on irradiation of a propylene carbonate/Cl2/N2 reaction mixture is shown in Fig. S2, trace (A) in the (ESI†). Although the spectrum looks very similar to that obtained in air, on closer inspection it is clear that another product is being formed that contains a carbonyl and hydroxyl entity. The product has been positively identified as acetic acid (CH3C(O)OH), a reference spectrum of which is shown in Fig. S2,† trace (B). The residual product spectrum which results on subtracting acetic acid from the product spectrum shown in Fig. S2,† trace (A) is shown in trace (C). The resulting spectrum is virtually identical with the product spectrum obtained in air and is attributed to the formation of acetyl formyl carbonate.
Fig. S3† shows the concentration–time profiles for the decay of propylene and the formation of acetic acid in N2. Also shown are the profiles for HC(O)OH and CO which were also formed. In N2 the yield of acetic acid was (42 ± 3)%. We have examined the formation of acetic acid as a function of the O2 partial pressure in the reaction system and the results are shown in Fig. S4.† It can be seen that the yields falls from ∼42% in N2 to zero by an O2 partial pressure of ∼20 Torr. Unfortunately we have no means of estimating just how large the trace levels of O2 are for the experiments performed in N2 but they are obviously sufficiently large that a significant fraction of the reaction leads to formation of acetyl formyl carbonate via the pathways outlined in Schemes 2 and 3.
We propose that the process leading to the formation of acetic acid at low O2 partial pressures involves an alternative reaction pathway for the alkoxy radical formed through H-atom abstraction from the tertiary carbon in propylene carbonate. We suggest that the process involves a 1,3-hydrogen shift from the methylene group to alkoxy oxygen as shown in Scheme S1 in the ESI.† The newly formed radical can undergo peroxy–peroxy reactions and eventually decompose to form acetic acid, CO2 and HO2 radicals. At present this is the only potentially viable route to the formation of acetic acid which we can think of. It is not possible to tell from the experiments whether the H-shift is thermal or photochemical.
Conclusions
Rate coefficients have been determined for the reaction of OH radicals and Cl atoms with propylene carbonate at room temperature and atmospheric pressure. Using an hydroxyl radical concentration of [OH] = 2 × 106 radicals cm3 (ref. 31) in combination with the OH rate coefficient determined in this work gives a tropospheric lifetime for propylene carbonate, with respect to reaction with OH, of around 24 days. The product study indicates that the main product of the atmospheric photooxidation of propylene carbonate will be acetyl formyl carbonate which does not appear to be particularly stable. Reaction of acetyl formyl carbonate with OH radicals will be very slow and since neither the carbonate nor anhydride entities in its structure absorb in the tropospheric solar actinic region (l > 290 nm)32,33 photolysis loss will also be negligible. Deposition on surfaces with formation of acetic and formic acids will probably constitute the main atmospheric fate of acetyl formyl anhydride. Therefore, the atmospheric degradation of propylene carbonate is likely to add to the environmental acidification burden close to point of its atmospheric in situ production.Acknowledgements
The authors thank Hüttenes-Albertus Chemische Werke GmbH, Düsseldorf, Germany for supplying propylene carbonate for the experiments.References
- H.-J. Buysch, Carbonic Esters, in Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Wiley-VCH, 2005. DOI:10.1002/14356007.a05_197.
- D. Stoye, Solvents, in Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Wiley-VCH, 2005. DOI:10.1002/14356007.a24_437.
- http://www.cosmeticsinfo.org/ingredient/propylene-carbonate-0#sthash.cBf4lYCY.dpuf .
- K. Nasirzadeh, R. Neueder and W. Kunz, J. Chem. Eng. Data, 2005, 50, 26–28 CrossRef CAS.
- https://pubchem.ncbi.nlm.nih.gov/compound/propylene_carbonate#section=Ecological-Information .
- B. J. Finlayson-Pitts and J. N. Pitts, J. Chemistry of the Upper and Lower Atmosphere, Academic Press, 1999 Search PubMed.
- W. M. Meylan and P. H. Howard, Chemosphere, 1993, 26, 2293–2299 CrossRef CAS.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353–356 CrossRef CAS.
- B. J. Finlayson-Pitts, M. J. Ezell and J. N. Pitts, Nature, 1998, 337, 241–244 CrossRef.
- A. A. P. Pszenny, E. V. Fischer, R. S. Russo, B. C. Sive and R. K. Varner, J. Geophys. Res., 2007, 112, D10S13 CrossRef.
- J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander and S. S. Brown, Nature, 2010, 464, 271–274 CrossRef CAS PubMed.
- L. H. Mielke, A. Fugeson and H. D. Osthoff, Environ. Sci. Technol., 2011, 45, 8889–8896 CrossRef CAS PubMed.
- G. J. Phillips, M. J. Tang, J. Thieser, B. Brickwedde, G. Schuster, B. Bohn, J. Lelieveld and J. N. Crowley, Geophys. Res. Lett., 2012, 39, L10811, DOI:10.1029/2012GL051912.
- T. P. Riedel, G. M. Wolfe, K. T. Danas, J. B. Gilman, W. C. Kuster, D. M. Bon, A. Vlasenko, S.-M. Li, E. J. Williams, B. M. Lerner, P. R. Veres, J. M. Roberts, J. S. Holloway, B. Lefer and S. S. Brown, Atmos. Chem. Phys., 2014, 14, 3789–3800 Search PubMed.
- G. Sarwar, H. Simon, J. Xing and R. Mathur, Geophys. Res. Lett., 2014, 41, 4050–4058, DOI:10.1002/2014GL059962.
- I. Barnes, K. H. Becker and N. Mihalopoulos, J. Atmos. Chem., 1994, 18, 267–289 CrossRef CAS.
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- E. S. C. Kwok and R. Atkinson, Atmos. Environ., 1995, 29, 1685–1695 CrossRef CAS.
- I. Barnes, S. Kirschbaum and J. M. Simmie, J. Phys. Chem. A, 2014, 118, 5013–5019 CrossRef CAS PubMed.
- S. Kirschbaum, Diploma thesis, University of Wuppertal, Germany, 2008.
- S. M. Aschmann and R. Atkinson, Int. J. Chem. Kinet., 1995, 27, 613–622 CrossRef CAS.
- J.-H. Xing, K. Takahashi, M. D. Hurley and T. J. Wallington, Chem. Phys. Lett., 2009, 474, 268–272 CrossRef CAS.
- P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat and F. Zabel, Atmos. Environ., Part A, 1992, 26, 1805–1964 CrossRef.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules, Chapman and Hall, London, vol. 2, 1978 Search PubMed.
- L. J. Bellamy, Advances in Infrared Group Frequencies, in The Infrared Spectra of Complex Molecules, Chapman and Hall, New York, vol. 2, 1980 Search PubMed.
- J. J. Orlando, L. T. Iraci and G. S. Tynall, J. Phys. Chem. A, 2000, 104, 5072–5079 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Chem. Rev., 2003, 103, 4657–4689 CrossRef CAS PubMed.
- J. G. Calvert, R. G. Derwent, J. J. Orlando, G. S. Tyndall and T. J. Wallington, Mechanisms of Atmospheric Oxidation of the Alkanes, Oxford University Press, New York, 2008 Search PubMed.
- R. Hein, P. J. Crutzen and M. Heimann, Global Biogeochem. Cycles, 1997, 11, 43–76 CrossRef CAS.
- M. Bilde, T. E. Møgelberg, J. Sehested, O. J. Nielsen, T. J. Wallington, M. D. Hurley, S. M. Japar, M. Dill, V. L. Orkin, T. J. Buckley, R. E. Huie and M. J. Kurylo, J. Phys. Chem. A, 1997, 101, 3514–3525 CrossRef CAS.
- The MPI-Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest: www.uv-vis-spectral-atlas-mainz.org.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21952g |
| This journal is © The Royal Society of Chemistry 2016 |