
Theoretical study of the non linear optical properties of alkali metal (Li, Na, K) doped aluminum nitride nanocages
Mariaa,
Javed Iqbalb and
Khurshid Ayub*a
aDepartment of Chemistry, COMSATS Institute of Information Technology, Abbottabad, Pakistan 22060. E-mail: Khurshid@ciit.net.pk; Fax: +92-992-383441; Tel: +92-992-383591
bDepartment of Chemistry, University of Agriculture, Faisalabad, Pakistan 22060
First published on 20th September 2016
Abstract
The effect of alkali metal (Li, Na, and K) doping in aluminum nitride (Al12N12) nanocages is studied through density functional theory (DFT) methods. Six new stable compounds of M@Al12N11 and M@Al11N12 are designed theoretically where alkali metal replaces an atom (Al/N) of a nanocage. The stability of the doped nano-cages is evaluated through binding energy calculations. Doping alkali atom M (M = Li, Na, K) into a nanocage significantly reduces the band gap (HOMO–LUMO gap). Polarizability and first hyperpolarizability are calculated using long range separated methods to evaluate the non-linear optical (NLO) properties of these doped systems. The hyperpolarizability of MAl12N11 nanocages is much higher than that of M@Al11N12 nanocages. The higher hyperpolarizability of M@Al12N11 nanocages is believed to arise from participation of excess diffuse electrons, revealed from PDOS.
Introduction
Over last few decades, significant efforts have been made to design novel materials exhibiting a large non-linear optical (NLO) response due to their versatile potential applications in photonics and opto-electronic devices.1–10 In view of designing high performance materials, a number of effective strategies have been proposed that can cause dramatic improvement in the NLO properties of a system (both organic and inorganic).11–22For organic materials the concept of extended conjugation combined with an enhanced electron push–pull mechanism has been a successful method of enhancing NLO response. In this approach, an electron donor and an acceptor fragment are carefully chosen and connected through π conjugation in a molecule.11,14,15 Different high performance NLO materials were designed using this strategy such as donor–acceptor-π bridge frameworks.13,16 Another approach for increasing NLO response of a system is to design metal–ligand frameworks in which transition metal is incorporated in organic molecules.23–25 Transitions caused by charge transfer from metal to ligand were exploited in this strategy. There are several other materials in which the same charge transfer transitions are exploited such as “octupolar materials”20,26 and “X-type chiral π-conjugated oligomers”.21
Recently, introduction of diffuse excess electrons in a system (organic/inorganic) has emerged as a proficient strategy for enhancement of NLO properties.27–33 Systems that contain diffuse excess electrons have considerably larger β0 (hyperpolarizability) value. Alkali metal atoms are excellent candidate for delivering diffuse excess electrons. Excess electrons from alkali metals can lower the excitation energy and hence play a decisive role for increasing β0 and enhanced NLO response.30,32,34–36 Different molecules such as calix[4]pyrrole,30 B4H10 basket,33 HCN trimer,28 π-conjugated aromatic rings like benzene, thiophene27 and most recently, AlN nanocages have been studied in this context.37 Interaction of these systems was studied with alkali metals (Li, Na, K), and it was found that valence electrons of alkali can be pushed out to generate diffuse excess electrons due to which excitation energy decreases which result in remarkably enhanced β0.
For last few years, nano structures such as fullerene hollow nanocages of elements other than carbon have gained much attraction due to their unique optical/electronic properties.38–43 Group III–V nitrides are among most promising nanocages.44,45 From group III nitrides, AlN nanocage has versatile applications in nanoscience due to its thermodynamic stability, low electron affinity and high thermal conductivity 42,46. A study by Wu et al., on the stability of aluminum nitride nanocages (n = 2–41) revealed that (AlN)12 nanocage is energetically the most stable one, and can be considered as an ideal nanocage. sp2 hybridization of group III metal atom and nitrogen is responsible for this stability.47 Al12N12 nanocages has been studied for its potential applications in hydrogen storage and in gas sensing. According to Wang et al., each aluminum atom in cage can adsorb one hydrogen molecule but when the nanocage is decorated with nickel, this adsorption capacity increases several times. Ni incorporated AlN nanocage can adsorb up to 20 molecules of hydrogen outside and inside the nanocage.48 NO adsorption on aluminum nitride nanocage was also studied and it was revealed that the nanocage in presence of CO can selectively adsorb/detect NO molecules.42 Beheshtian showed that fluorine atom can be adsorbed on aluminum site of nanocage, and this fluorination can significantly alter the electronic properties of nanocage to p-type semiconductor.49 Al12N12 has been studied for NLO response when doped with alkali metal. Alkali metal incorporation can be done in many ways, such as placing alkali metal atom at the surface of the nanocage, encapsulation of alkali metal inside nanocage and replacement of any atom of nanocage by alkali atom. Surface doping on Al12N12 nanocage enhances the first hyperpolarizability by several orders of magnitude.37 Second approach is recently used by Zeinab et al., for group III nitrides where alkali metal was encapsulated in the nanocage that results in larger first hyperpolarizability of the system.45 Nevertheless, a third approach of doping is yet to be explored where alkali atom replaces any atom of the nanocage.
In current study, we investigate the NLO properties of inorganic nano clusters by doping them with alkali metal atom. This work mainly aims to investigate the impact of alkali metals (Li, Na, K) doping on NLO properties of (AlN)12 nanocage. Here, in this work, the replacement of aluminum or nitrogen atom of nanocage with alkali atom gives rise to two types of structures, M@Al12N11 (where nitrogen is replaced with alkali metal) and M@Al11N12 (where aluminum is replaced). Main objective of our work is to investigate thoroughly the influence of alkali metal doping at different sites on its optical and electronic properties. The results are quite useful for designing of new materials for their potential applications in electronics and high-performance NLO materials.
Methodology
Gaussian 09 suit of programs is used for all calculations.50 All the geometric structures are optimized at B3LYP/6-31G(d, p) level of theory. No imaginary frequency is found for these structures which confirms them as true minima. Stability of undoped and doped systems was evaluated by binding energies and cohesive energy:(Cohesive)
Undoped system
| Ecoh = EAlN − (12EAl + 12EN) |
N-site doped system
| Ecoh = EAlNM − (12EAl + 11EN + 1EM) |
Al-site doped system
| Ecoh = EAlNM − (11EAl + 12EN + 1EM) |
(Adsorption)
For M@Al12N11
| Ebind = EAlNM − (EAl12N11 + EM) |
For M@Al11N12
| Ebind = EAlNM − (EAl11N12 + EM) |
Polarizability and first hyperpolarizability calculations are also carried out on long range correlated methods such as CAM-B3LYP/6-311+g(d) and LC-BLYP/6-311+G(d) level of theory.51–55 CAM-B3LYP/6-311+g(d) and LC-BLYP/6-311+G(d) are the best reported level of theory for the calculation of first hyperpolarizability.45 However, a recent report shows that CAM-B3LYP slightly overestimate hyperpolarizabilities.36,56,57 Our results (vide infra) are in accordance with statement.
First hyperpolarizability and mean polarizability is denoted as
Polarizability
| α = 1/3(αxx, αyy, αzz) |
First hyperpolarizability
| β0 = [(βxxx, βxyy, βzzz)2 + (βyyy, βyzz, βyxx)2 + (βzzz, βzxx, βzyy)2]1/2 |
Moreover, for calculation of crucial excited states of all the structures TD-DFT (time dependent DFT) calculations are executed on CAM-B3LYP/6-311+g(d) level of theory. Same basis set is employed for the calculation of dipole moment of excited states.
Density of states of all the structures are plotted by using GaussSum and partial density of states (PDOS) are generated through multiwfn software.58
Results and discussion
All alkali doped aluminum nitride nano cages (M@Al12N12) geometries were optimized without any symmetry constraints through DFT method. Two sets of geometries were optimized namely, M@Al12N11 and M@Al11N12 (M is alkali metal). These two series differ according to doping site in nanocage. In M@Al12N11 series, nitrogen is replaced with alkali metal whereas, aluminum is replaced with alkali atom in M@Al11N12 (Fig. 1). For the assessment of stability of both series, binding and cohesive energies were calculated and compared with pure Al12N12 nanocage. Pure Al12N12 nanocage (Ecoh of 3136 kcal mol−1) is more stable than metal doped Al12N12 nanocage. It was observed that stability of doped nanocages decreased slightly with increase in atomic number of alkali metal. In M@Al12N11 series, Li@Al12N11 has highest cohesive energy value of 2926 kcal mol−1 which decreases very slightly for Na@Al12N11 (2914 kcal mol−1) and K@Al12N11 (2912 kcal mol−1). Similarly M@Al11N12 series also showed a very little difference in cohesive energy values among different doped nanocages. Li@Al11N12 has cohesive energy of 3020 kcal mol−1 which is higher than 2998 kcal mol−1 for Na@Al11N12 and 2995 kcal mol−1 for K@Al11N12. M@Al11N12 series showed higher cohesive energy than M@Al12N11 series.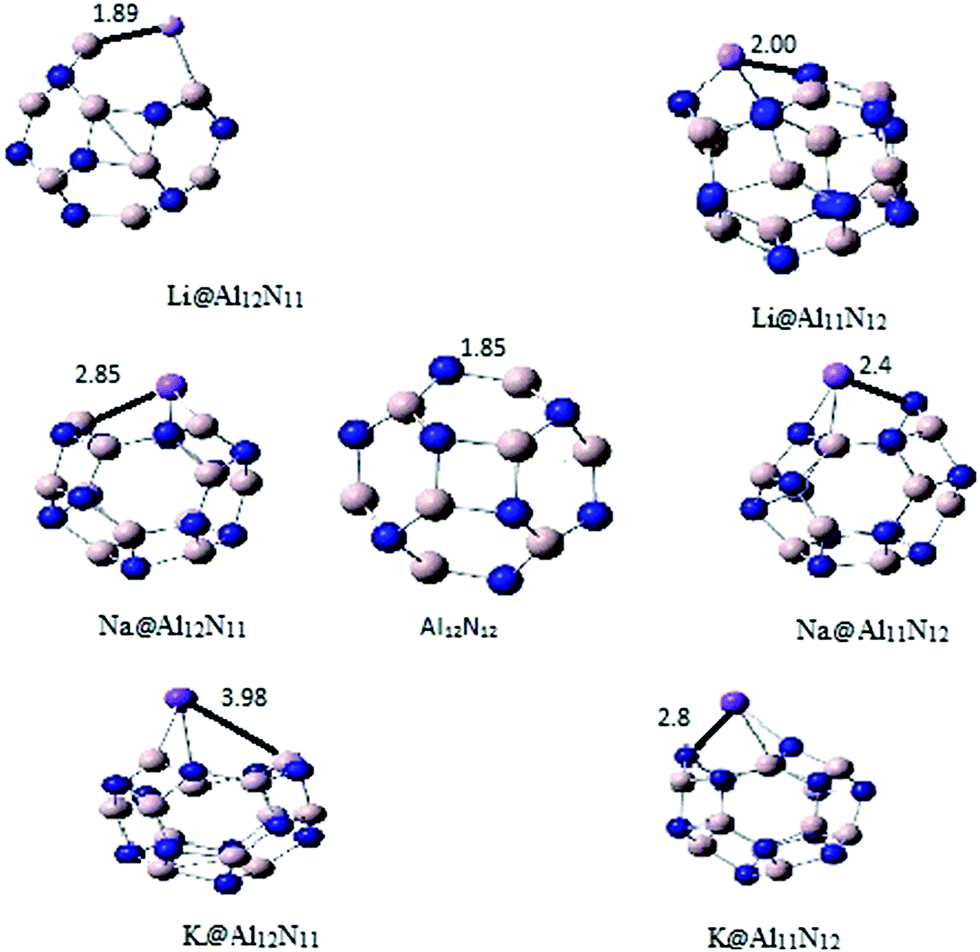 | ||
| Fig. 1 Optimized structures of undoped and alkali doped aluminum nitride nanocages (all the bond lengths are in angstrom (Å)). | ||
Furthermore, binding energy (Ebind) values are also coherent with cohesive energy values where, Li@Al12N11 has the highest adsorption energy value of −45 kcal mol−1 among M@Al12N11. Na@Al12N11 has adsorption energy of −33 kcal mol−1 which decreases to −31 kcal mol−1 for K@Al12N11. Similarly, Li@Al11N12 has the highest value of Ebind (−89 kcal mol−1) in M@Al11N12 series. Na@Al11N12 has binding energy value of −67 kcal mol−1, and it decrease further for K@Al11N12 (−64 kcal mol−1). The high binding energy of M@Al11N12 can be attributed to the strong binding of nitrogen with alkali metal in doped nanocages. Interaction of nitrogen with alkali atom results in strong binding interactions. While in M@Al12N11 series, bonds are formed between alkali metal and aluminum atom which have weak binding interactions than M@Al11N12.
Bond length of M–Al and M–N (M = alkali metal atom) in pure Al12N12 is 1.84 Å, which shows a constant increase with increase in atomic number of doped alkali metal. M–Al bond in Li@Al12N11 is 2.5 Å which is less than (2.85 Å) for Na@Al12N11 whereas, K@Al12N11 has highest bond length of 4.0 Å. Likewise, for M@Al11N12 series, M–N bond also showed significant increase in bond length for heavier alkali metals. K@Al11N12 showed bond length of about 2.8 Å which is greater than 2.4 Å for Na@Al11N12. Li@Al11N12 has lowest bond length of 2.07 Å. M@Al12N11 series displayed larger increment in bond length than M@Al11N12 series (Table 1).
| Properties | Pure Al12N12 | MAl12N11 | MAl11N12 | ||||
|---|---|---|---|---|---|---|---|
| Li | Na | K | Li | Na | K | ||
| Symmetry | Th | C1 | C1 | C1 | C1 | C1 | C1 |
| Bond length (Å) | 1.85 | 1.98 | 2.85 | 3.98 | 2 | 2.4 | 2.8 |
| Charge q | 0.517 | 0.561 | 0.761 | 0.744 | 0.496 | 0.788 | |
| % P | 54 | 57 | 61 | 72 | — | — | |
| % S | 45 | 41 | 37 | 27 | — | — | |
| Ecoh (kcal mol−1) | 3136 | 2926 | 2914 | 2912 | 3020 | 2998 | 2995 |
| Ebind (kcal mol−1) | — | −45 | −33 | −31 | −89 | −67 | −64 |
| Eg (eV) | 3.9 | 1.39 | 1.78 | 1.74 | 2.9 | 2.8 | 2.7 |
NBO analysis was carried not only to find out % S and % P character in all M–Al and M–N bonds but also to analyze charge transfer in nanocage. In all doped nanocages considered here, alkali metal was found to be more electropositive than remaining nanocage. Charge is transferred from alkali atom to the nanocage causing the later more electron rich. Hence, an assumption was made that s valence electrons were transferred from alkali atom to the nanocage forming alkali metal more electropositive. NBO analysis showed that each bond (vide infra) (connecting alkali to nanocage atom) has more % P character than % S character which is assumed to be responsible for bond lengthening of M–Al and M–N than all other bonds in nanocage. % P character in orbitals was seen to contribute more in M@Al12N11 series than in M@Al11N12 series which leads to increase in bond length in the former series due to very weak bonding (long bond) between alkali metal and nanocage. For M@Al12N11 series, % P character increases for aluminum atom with increase in atomic number of bonded alkali atom. K@Al12N11 has highest % P character of 61% for aluminum atom (Table 1). The % P and % S character could not be found for Na@Al11N12 and K@Al11N12 nanocages. This is probably due to very weak interactions of alkali metal with the nanocage.
Electronic properties of all considered doped and undoped nanocages were examined to explore the effect of doping. For this purpose, we have studied frontier molecular orbitals, band gap and density of states. Energies of HOMO and LUMO as well as band gap (Eg) were calculated for both series. Pure Al12N12 has a large band gap of 3.9 eV which is a barrier in the way of its applications in electronic devices. It was observed that electronic properties for doped nanocages were much different than undoped nanocages since a significant decrease was noticed in HOMO–LUMO gap for all doped Al12N12 nanocages (Table 1).
Band gap is lowered down to 1.39 eV for Li@Al12N11 which slightly increases to 1.78 eV for Na@Al12N11. Eg for K@Al12N11 is 1.74 eV which is slightly different from band gap of Na@Al12N11. On the other hand, M@Al11N12 series showed decreasing trend of band gap with increase in atomic number of alkali metal hence lowest value was found for K@Al11N12 (2.7 eV). Na@Al11N12 and Li@Al11N12 showed band gap of 2.8 eV and 2.9 eV, respectively. M@Al12N1 series showed much decrease in band gap than M@Al11N12 series (Fig. 2, Table 1). Comparison of these Eg values with original band gap value of pure AlN (3.9 eV) suggests larger first hyperpolarizability of doped nanocages.
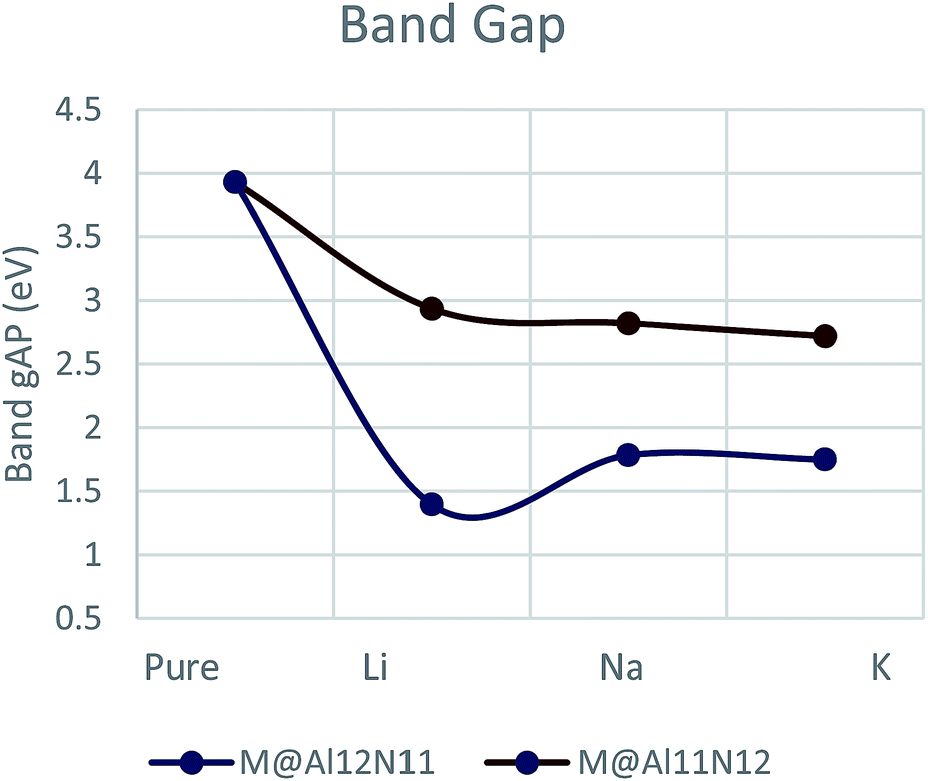 | ||
| Fig. 2 Graphical representation of band gap. | ||
Decrease in band gap in alkali doped AlN nano cages is assumed to arise from the formation of a new high energy level above the original HOMO of pure nanocage. This new HOMO is located between old HOMO and LUMO which minimizes the gap between them. In case of pure AlN nano cage, LUMO appeared at −2.54 eV and HOMO was present at −6.46 eV. HOMO in Li@Al12N11 were shifted at −5.09 eV from −5.97 eV (Fig. 3). For Na@Al12N11 and for K@Al12N11 energy levels were shifted to −4.47 eV and −4.16 eV from original value of −5.77 eV and −5.53 eV, respectively. Likewise, M@Al11N12 series have fermi levels changed from-5.66 eV to −5.45 eV for Li@Al11N12. Fermi levels of Na@Al11N12 and K@Al11N12 were changed to −5.20 eV and −4.96 eV from −5.34 eV and −5.01 eV, respectively (Fig. 3).
 | ||
| Fig. 3 TDOS showing energy levels of doped aluminum nitride nanocage. | ||
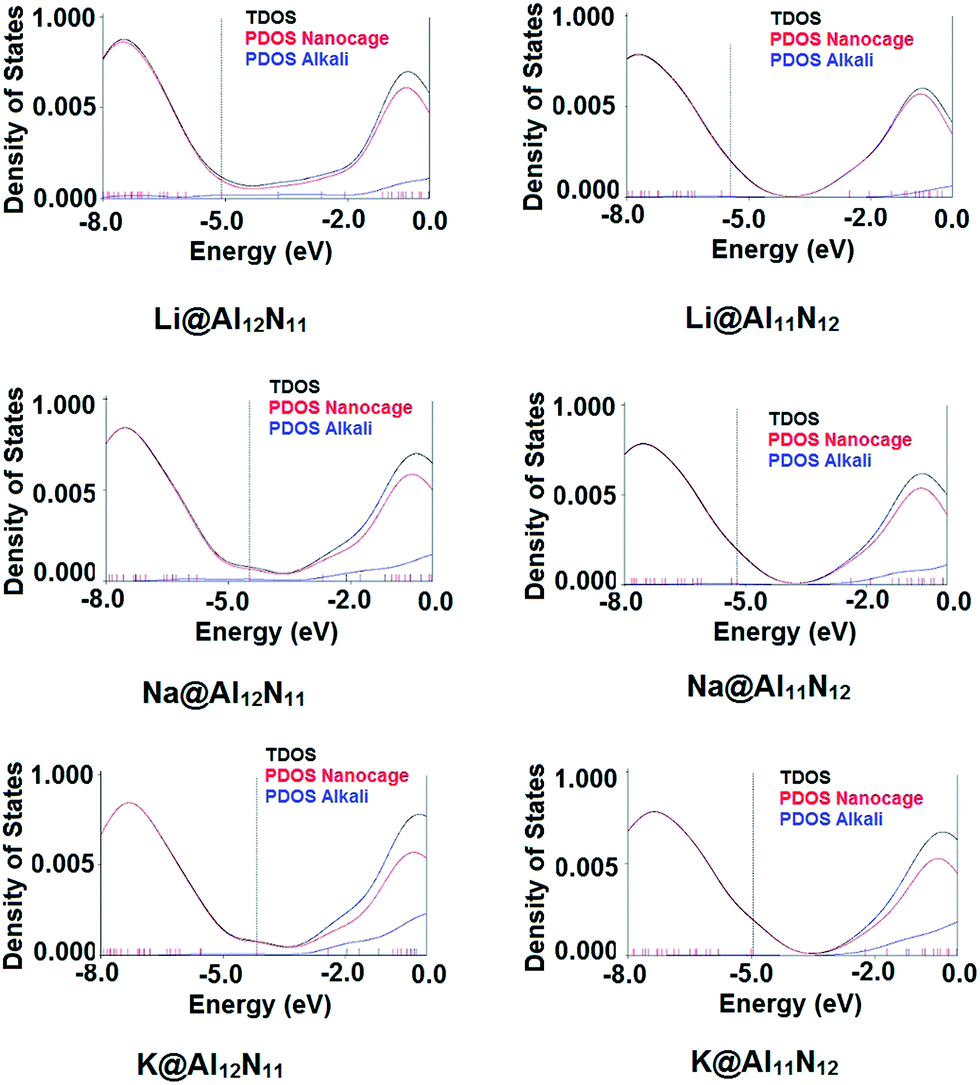
To better understand the electronic properties, total density of states (TDOS) were plotted for both pure and doped AlN nanocages. For M@Al12N11 series, TDOS indicated that doping the nanocage with alkali metal from Li to K results in formation of new levels. These new energy levels resides at higher energy than old HOMO hence, constitute the new HOMO. Diffuse excess electrons from alkali metal atom were transferred to nanocage due to which new level was generated and therefore, band gap is lowered in doped AlN nanocages. Same is the case for M@Al11N12 series, where TDOS clearly point to the formation of new HOMO. Old HOMO serves as HOMO-1 in doped nano cages.
In all doped nanocages, LUMO was present at almost same energy value as of original LUMO (−2.5 eV) however, in case of Li@Al12N11, the new LUMO also appeared at much lower energy value (−3.70 eV). Hence, Li@Al12N11 has lowest band gap in all doped AlN nano cages. It is obvious from the results that presence of diffuse excess electrons in a system can make a great change in its conducting properties through formation of new energy levels and as a result lowering the band gap. To confirm the involvement of alkali's valence electrons in formation of new HOMO, PDOS were plotted. In Fig. 4, PDOS of alkali atom doped structures are present which confirms the contribution of alkali metal in generating new HOMO level.
 | ||
| Fig. 4 PDOS of doped structures (dashed line corresponds to HOMO). | ||
Earlier studies confirmed that the presence of diffuse excess electrons can cause large NLO response of a system.1,5,14,37,45 Presence of these electrons can considerably change polarizability and first hyperpolarizability of a system.
In this work we computed polarizability and first hyperpolarizability of pure and alkali doped AlN nanocages. Polarizability (α) of pure Al12N12 was found to be 287 au, which significantly increased for all considered nano cages. In M@Al12N11 series, increasing trend of polarizability with increase in atomic number of alkali was seen hence, K@Al12N11 showed highest value of 397 au. Li@Al12N11 has the lowest α of 350 au while, Na@Al12N11 has α value of 374 au which is larger than Li@Al12N11 and smaller than K@Al12N11. On the other hand, M@Al11N12 series showed increasing trend of polarizability however, very slight increase in α value of pure nano cage was observed. Li@Al11N12 has α of 288 au which increases for Na@Al11N12 (294 au) and further increases to 301 au for K@Al11N12. Here, we noticed that M@Al12N11 series showed much increase in α than M@Al11N12 series.
Much attention is given to first hyperpolarizability (β0) due to interesting change observed in its value for alkali doped AlN nanocages. First Hyperpolarizability of pure Al12N12 was negligible however, β0 is considerably increased for doped nanocages. Two different long range corrected methods were used to calculate hyperpolarizability (CAM-B3LYP and LC-BLYP).
CAM-B3LYP
With CAM-B3LYP method, for M@Al12N11 series, β0 was found to be 2.5 × 103 au (2561.6 au) for Li@Al12N11 which increases to 4.8 × 103 au (4834.6 au) for Na@Al12N11 and 9.1 × 104 (9134.7 au) for K@Al12N11. A relatively different trend is observed for M@Al11N12 series, where highest β0 is calculated for Na@Al11N12 that is 7.7 × 102 au (779.4 au). β0 for lighter alkali Li@Al11N12 and heavier alkali K@Al11N12 are 4.2 × 102 au (428.8 au) and 7.1 × 102 au (716.2 au), respectively. M@Al12N11 series, have larger first hyperpolarizability than M@Al11N12 series.LC-BLYP
Hyperpolarizability at LC-BLYP for M@Al12N11 and M@Al11N12 follows the same trend as for CAM-B3LYP. However, the calculated values at LC-BLYP are slightly lower than CAM-B3LYP. This is consistent with the literature that CAM-b3lyp overestimate the hyperpolarizability value.56,57 In M@Al12N11 series, Li@Al12N11 has lowest β0 of 2.1 × 103 au (2100.3 au), which increases to 4.6 × 103 au (4603.5 au) for Na@Al12N11. K@Al12N11 has largest value of 6.6 × 103 au (6615.8 au) in the series. For M@Al11N12 series, Na@Al11N12 has largest β0 of 4.8 × 102 au (487.6 au) whereas, Li@Al11N12 has lowest value of 2.6 × 102 au (263.2 au). K@Al11N12 was found to have β0 of 3.9 × 102 au (394.1 au). It is assumed that in former series bonding interactions were relatively weak due to larger distance present among alkali and its bonded atom. This greater distance may create larger dipole moment hence, give rise to larger β0 for M@Al12N11. It is depicted here that in M@Al12N11 series, ionization energy plays its role due to which monotonic behavior is seen.Both the method showed significant increase and trends in hyperpolarizability of considered compounds however, values obtained from CAM-B3LYP method were greater than values obtained from LC-BLYP method (Fig. 5).
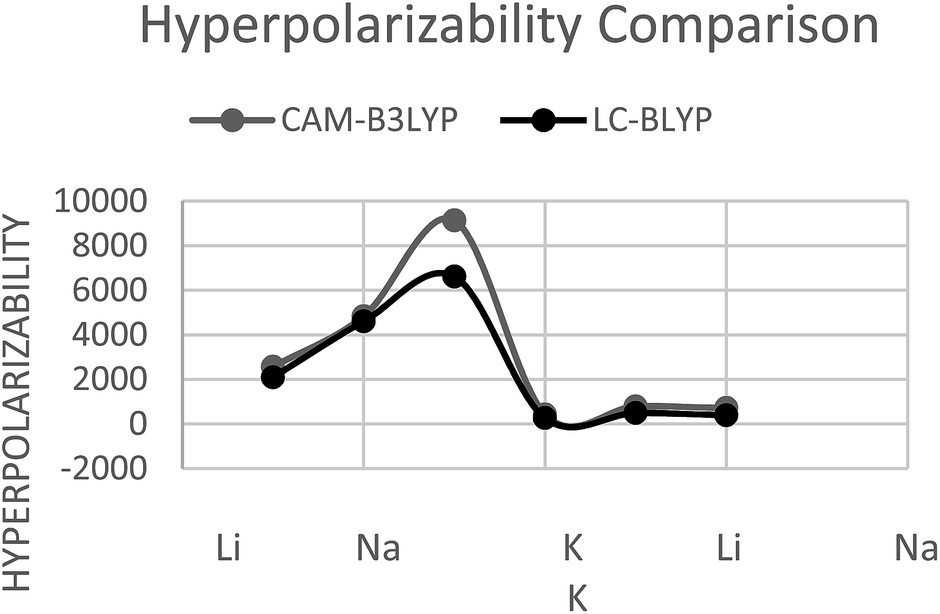 | ||
| Fig. 5 Hyperpolarizability comparison on two different methods. | ||
For deep understanding of this tremendous increase in first hyperpolarizability due to doping of alkali metal in AlN nano cage, “two level model” was considered. According to two level model
| β0 ≈ Δμ × f0/ΔE3 |
Monotonic dependency
Earlier work shows that heavier alkali atom have larger effect on NLO response of system.27,33,56,59 Ionization potential of alkali decreases with increase in atomic number of alkali therefore, valence electrons have weak interactions with metal center for larger atomic number and are easy to push out.Hyperpolarizability of a system can be decided by several factors. For example, ionization potential can affect the hyperpolarizability if diffuse excess electrons are present as the only factor subsequently a monotonic behavior is predicted. This is attributed to the fact that diffuse excess electrons are related to ionization potential and with increase in the atomic number of alkali ionization potential also increases. In our results M@Al12N11 have shown monotonic increase in hyperpolarizability values which is in agreement with the trend discussed above.
There are also reports in literature which supports our results for monotonic dependency of hyperpolarizability on atomic number.56,60 PDOS are also plotted for sustenance of the results.
Another factor that may play its role in deciding hyperpolarizability is the interaction distance (distance between alkali atom and the system). Shorter distance leads to high hyperpolarizability since the interaction of alkali atom and the system is stronger whereas, longer distance reverse the trend of hyperpolarizability. Thus, if interaction distance is only factor for deciding hyperpolarizability then a decrease in hyperpolarizability is estimated with increase in atomic number of alkali. There are also reports in literature which shows this type of trend.37,57
Nevertheless, the behavior observed for our calculated hyperpolarizability values of M@Al11N12 is unmonotonic, which proposes that not a single factor from the above discussed factors is playing its part in the series. Hence, it is anticipated that both the factors together are affecting this trend. An increase in hyperpolarizability of Na@Al11N12 over Li@Al11N12 may be due to ionization potential and decrease in hyperpolarizability of K@Al11N12 than Na@Al11N12 is probably due to interaction distance.
Moreover, β0 was also calculated from above mentioned ‘two level model’ expression and the results are given in Table 2.
| Properties | Pure Al12N12 | MAl12N11 | MAl11N12 | ||||
|---|---|---|---|---|---|---|---|
| Li | Na | K | Li | Na | K | ||
| α (au) | 284 | 350 | 374 | 397 | 282 | 294 | 301 |
| β (CAM-B3LYP) | 2.5 × 103 | 4.8 × 103 | 9.1 × 103 | 4.2 × 102 | 7.7 × 102 | 7.1 × 102 | |
| β (LC-BLYP) | 2.1 × 103 | 4.6 × 103 | 6.6 × 103 | 2.6 × 102 | 4.8 × 102 | 3.9 × 102 | |
| f0 | 0 | 0.0596 | 0.3099 | 0.3784 | 0.0169 | 0.0169 | 0.0155 |
| ΔE (eV) | 4.6 | 5.5 | 5.3 | 6.7 | 5.4 | 5.2 | |
| Δμ (eV) | 4.8 | 0.5 | 5.6 | 0.5 | 1.10 | 1.4 | |
| Δμ × f0/ΔE3 | 3261 | 1033 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 795 795 |
31 | 138 | 171 | |
| CT | H → L + 1 | H → L + 4 | H → L + 6 | H − 5 → L | H − 1 → L | H → L | |
| H → L + 3 | H → L + 10 | H − 3 → L | H − 1 → L + 3 | H − 1 → L | |||
We analyzed all components of β0 in two level model to realize which component has the highest contribution. ΔE, does not yield the trend as observed with calculated β0. For example, Na doped nanocage (Na@Al12N11) showed higher ΔE than other alkali metals (Li, K). Then, f0 (largest oscillator strength) is analyzed for M@Al12N11. f0 shows monotonic behavior where value for K@Al12N11 is higher (0.3784) than both Na@Al12N11 (0.3099) and Li@Al12N11 (0.0596). In M@Al12N11, f0 shows a trend consistent with β0 which suggests that oscillator strength is playing a key role in deciding the trend for hyperpolarizability.
For M@Al11N12 series, ΔE and f0 are also analyzed which showed unmonotonic trend of both factors. Both ΔE and f0 decrease with increase in atomic number. ΔE for Li@Al11N12 (6.7 eV) is higher than Na@Al11N12 (5.4 eV) and K@Al11N12 (5.2 eV). Likewise, Li@Al11N12 and Na@Al11N12 have almost same value of f0 0.0169 which decreased for K@Al11N12 (0.0155). These results again suggests that f0 is the key factor in deciding the trends of hyperpolarizability.
Above discussion clearly depicted that f0 can be conclusive factors for explaining the particular trend of first hyperpolarizability for M@Al12N11 series. Consequently, dipole moments (Δμ) of ground state and excited state of crucial transitions were calculated (Table 2).
Conclusion
In the present study six new alkali doped aluminum nitride nanocages are designed and studied through DFT where, alkali metal replaces either aluminum or nitrogen atom (M@Al12N11 and M@Al11N12). % S and % P character for alkali metal–atom (M–Al/M–N) bond was calculated and it was found that contribution of % P is more than % S character which results in lengthening of the bond. Band gap (Eg) is narrowed significantly from 3.9 eV for pure Al12N12 nanocage to 1.39 eV for Li@Al12N11. For other nanocages band gap lie in the range of 1.74 eV to 2.9 eV. Valence electrons from alkali atom are pushed out to form new energy levels near HOMO. This new HOMO results in lowering the gap between HOMO and LUMO.Moreover, introduction of these diffuse excess electrons in M@Al12N11 and M@Al11N12 are responsible for increasing hyperpolarizability of system. Hyperpolarizability was calculated on long range corrected methods such as CAM-B3LP and LC-BLYP. β0 for our newly designed structures are increased several times where, K@Al12N11 has largest value of 9.1 × 103 au. M@Al12N11 series is found to have larger hyperpolarizability values than M@Al11N12. Two state model is used for estimation of first hyperpolarizability of optimized structures and results indicate that oscillator strength is a key factor in deciding the hyperpolarizability of the newly designed nanocages.
Acknowledgements
Author acknowledge financial support for the research of the manuscript from Higher Education Commission (HEC) Grant number 1899, 2469, 2981. The author acknowledge full support from Higher Education Commission, COMSATS Institute of Information Technology, Abbottabad, and University of Agriculture, Faisalabad.References
- H. L. Xu, R. L. Zhong, S. Muhammad, J. Zhang and Z. M. Su, J. Mater. Chem., 2012, 22, 2196–2202 RSC.
- C. Tu, G. Yu, G. Yang, X. Zhao, W. Chen, S. Lia and X. Huang, Phys. Chem. Chem. Phys., 2014, 16, 1597–1606 RSC.
- S. Muhammad, H. Xu and Z. Su, J. Phys. Chem. A, 2011, 115, 923–931 CrossRef CAS PubMed.
- Y. Y. Hu, S. L. Sun, S. Muhammad, H. L. Xu and Z. M. Su, J. Phys. Chem. C, 2010, 114, 19792–19798 CAS.
- E. Shakerzdeh, E. Tahmasebi and H. R. Shamlouei, Synth. Met., 2015, 204, 17–24 CrossRef CAS.
- N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2003, 42, 2586–2617 CrossRef CAS PubMed.
- D. F. Eaton, Science, 1991, 253, 281–287 CAS.
- M. Blanchard-Desce, V. Alain, P. V. Bedworth, S. R. Marder, A. Fort, C. Runser, M. Barzoukas, S. Lebus and R. Wortmann, Chem.–Eur. J., 1997, 3, 1091–1104 CrossRef CAS.
- S. R. Marder, W. E. Torruellas, M. Blanchard-Desce, V. Ricci, G. I. Stegeman, S. Gilmour, J. L. Brédas, J. Li, G. U. Bublitz and S. G. Boxer, Science, 1997, 276, 1233–1236 CrossRef CAS PubMed.
- B. Champagne, A. Plaquet, F. Castet, L. Ducasse, E. Bogdan, V. Rodriguez and J. L. Pozzo, New J. Chem., 2009, 33, 1349–1356 RSC.
- D. J. E. Williams, ACS Symp. Ser., 1984, 233 Search PubMed.
- D. Jacquemin, B. Champagne and B. Kirtman, J. Chem. Phys., 1997, 107, 5076 CrossRef CAS.
- M. Schulz, S. Tretiak, V. Chernyak and S. Mukamel, J. Am. Chem. Soc., 2000, 122, 452–459 CrossRef CAS.
- S. Priyadarshy, M. J. Therien and D. N. Beratan, J. Am. Chem. Soc., 1996, 118, 1504–1510 CrossRef.
- D. M. Bishop, B. Champagne and B. Kirtman, J. Chem. Phys., 1998, 109, 9987 CrossRef CAS.
- D. Xiao, F. A. Bulat, W. Yang and D. N. Beratan, Nano Lett., 2008, 8, 2814–2818 CrossRef CAS PubMed.
- T.-G. Zhang, Y. Zhao, I. Asselberghs, A. Persoons, K. Clays and M. J. Therien, J. Am. Chem. Soc., 2005, 127, 9710–9720 CrossRef CAS PubMed.
- B. J. Coe, J. Fielden, S. P. Foxon, I. Asselberghs, K. Clays and B. S. Brunschwig, Inorg. Chem., 2010, 49, 10718 CrossRef CAS PubMed.
- J. Guthmuller, F. Zutterman and B. Champagne, J. Chem. Phys., 2009, 131, 154302 CrossRef PubMed.
- O. Maury, L. Viau, K. Senechal, B. Corre, J. P. Guegan, T. Renouard, I. Ledoux, J. Zyss and L. H. Bozec, Chem.–Eur. J., 2004, 10, 4454 CrossRef CAS PubMed.
- D. Cornelis, E. Franz, I. Asselberghs, K. Clays, T. Verbiest and G. Koeckelberghs, J. Am. Chem. Soc., 2011, 133, 1317 CrossRef CAS PubMed.
- S. Keinan, M. J. Therien, D. N. Beratan and W. Yang, J. Phys. Chem. A, 2008, 112, 12203 CrossRef CAS PubMed.
- G. de la Torre, P. Vázquez, F. Agulló-López and T. Torres, Chem. Rev., 2004, 104, 3723–3750 CrossRef CAS PubMed.
- C.-G. Liu, W. Guan, P. Song, L.-K. Yan and Z.-M. Su, Inorg. Chem., 2009, 48, 6548–6554 CrossRef CAS PubMed.
- N. Tancrez, C. Feuvrie, I. Ledoux, J. Zyss, L. Toupet, H. Le Bozec and O. Maury, J. Am. Chem. Soc., 2005, 127, 13474–13475 CrossRef CAS PubMed.
- S. H. Lee, J. R. Park, M. Y. Jeong, H. M. Kim, S. J. Li, J. Song, S. Ham, S. J. Jeon and B. R. Cho, ChemPhysChem, 2006, 7, 206 CrossRef CAS PubMed.
- G. Yu, X.-R. Huang, W. Chen and C.-C. Sun, J. Comput. Chem., 2011, 32, 2005–2011 CrossRef CAS PubMed.
- W. Chen, Z.-R. Li, D. Wu, R.-Y. Li and C.-C. Sun, J. Phys. Chem. B, 2005, 109, 601–608 CrossRef CAS PubMed.
- W. Chen, Z.-R. Li, D. Wu, Y. Li, R.-Y. Li and C.-C. Sun, J. Phys. Chem. A, 2005, 109, 2920–2924 CrossRef CAS PubMed.
- W. Chen, Z.-R. Li, D. Wu, Y. Li, C.-C. Sun, F. L. Gu and Y. Aoki, J. Am. Chem. Soc., 2006, 128, 1072–1073 CrossRef CAS PubMed.
- H. L. Xu, Z. R. Li, D. Wu, B. Q. Wang, Y. Li, F. L. Gu and Y. Aoki, J. Am. Chem. Soc., 2007, 129, 2967 CrossRef CAS PubMed.
- F. F. Wang, Z. R. Li, D. Wu, B. Q. Wang, Y. Li, Z. J. Li, W. Chen, G. T. Yu, F. L. Gu and Y. Aoki, J. Phys. Chem. B, 2008, 112, 1090 CrossRef CAS PubMed.
- S. Muhammad, H. Xu, Y. Liao, Y. Kan and Z. Su, J. Am. Chem. Soc., 2009, 131, 11833–11840 CrossRef CAS PubMed.
- W. Chen, Z. R. Li, D. Wu, Y. Li and C. C. Sun, J. Phys. Chem. A, 2005, 109, 2920 CrossRef CAS PubMed.
- W. Chen, Z. R. Li, D. Wu, F. L. Gu, X. Y. Hao, B. Q. Wang, R. J. Li and C. C. Sun, J. Chem. Phys., 2004, 121, 10489 CrossRef CAS PubMed.
- Z.-J. Li, F.-F. Wang, Z.-R. Li, H.-L. Xu, X.-R. Huang, D. Wu, W. Chen, G.-T. Yu, F. L. Gu and Y. Aoki, Phys. Chem. Chem. Phys., 2009, 11, 402–408 RSC.
- M. Niu, G. Yu, G. Yang, W. Chen, X. Zhao and X. Huang, Inorg. Chem., 2014, 53, 349–358 CrossRef CAS PubMed.
- D. L. Strout, J. Phys. Chem. A, 2000, 104, 3364 CrossRef CAS.
- R. X. Wang, D. J. Zhang and C. B. Liu, Chem. Phys. Lett., 2005, 411, 333 CrossRef CAS.
- M. Bertolus, F. Finocchi and P. Millié, J. Chem. Phys., 2004, 120, 9 Search PubMed.
- C. C. Fu, M. Weissmann, M. Machado and P. Ordejón, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 85411 CrossRef.
- J. Beheshtian, Z. Bagheri, M. Kamfiroozi and A. Ahmadi, J. Mol. Model., 2012, 2653–2658 CrossRef CAS PubMed.
- A. A. Peyghan, M. Pashangpour, Z. Bagheri and M. Kamfiroozi, Phys. E, 2012, 44, 1436 CrossRef CAS.
- A. K. Kandalam, M. A. Blanco and R. Pandey, J. Phys. Chem. B, 2001, 105, 6080–6084 CrossRef CAS.
- E. Tahmasebi, E. Shakerzadeh and Z. Biglari, Appl. Surf. Sci., 2016, 363, 197–208 CrossRef CAS.
- F. Zhang, Q. Wu, X. Wang, N. Liu, J. Yang, Y. Hu, L. Yu, X. Wang, Z. Hu and J. Zhu, J. Phys. Chem. C, 2009, 113, 4053–4058 CAS.
- H.-S. Wu, F.-Q. Zhang, X.-H. Xu, C.-J. Zhang and H. Jiao, J. Phys. Chem. A, 2003, 107, 204–209 CrossRef CAS.
- H. Wu, X. Fan and J.-L. Kuo, Int. J. Hydrogen Energy, 2012, 37, 14336–14342 CrossRef CAS.
- J. Beheshtian, A. A. Peyghan and Z. Bagheri, Appl. Surf. Sci., 2012, 259, 631–636 CrossRef CAS.
- M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, Z. Cheeseman, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, J. Tomasi, M. Millam, S. Dapprich, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. V. B. Tomasi, M. Cossi, R. Cammi and B. Mennucci, Gaussian 09, Rev C.01, Pittsburg, 2010 Search PubMed.
- S. Nénon, B. Champagne and M. I. Spassova, Phys. Chem. Chem. Phys., 2014, 16, 7083 RSC.
- M. B. Oviedo, N. V. Ilawe and B. M. Wong, J. Chem. Theory Comput., 2016, 12, 3593–3602 CrossRef PubMed.
- B. M. Wong and J. G. Cordaro, J. Chem. Phys., 2008, 129, 214703 CrossRef PubMed.
- B. Kirtman, S. Bonness, A. Ramirez-Solis, B. Champagne, H. Matsumoto and H. Sekino, J. Chem. Phys., 2008, 128, 114108 CrossRef PubMed.
- B. Champagne and D. M. Bishop, in Advances in Chemical Physics, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003, pp. 41–92 Search PubMed.
- W. Chen, Z.-R. Li, D. Wu, Y. Li, C.-C. Sun and F. L. Gu, J. Am. Chem. Soc., 2005, 127, 10977–10981 CrossRef CAS PubMed.
- W. Chen, G. Yu, P. Jin, Z.-R. Li and X.-R. Huang, J. Comput. Theor. Nanosci., 2011, 8, 2482–2487 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- L. Z. Kang, T. Inerbaev, B. Kirtman and F. L. Gu, Theor. Chem. Acc., 2011, 130, 727–737 CrossRef CAS.
- Z. J. Li, F. F. Wang, Z. R. Li, H. L. Xu, X. R. Huang, D. Wu, W. Chen, G. T. Yu, F. L. Gu and Y. Aoki, Phys. Chem. Chem. Phys., 2009, 11, 402 RSC.
| This journal is © The Royal Society of Chemistry 2016 |