
Synthesis and characterization of vinyl ester/inorganic layered material nanocomposites
Tsung-Yen Tsai*ab,
Naveen Bunekara,
Chein-Hsiang Yena and
Yu-Bing Lina
aDepartment of Chemistry, Chung-Yuan Christian University, Chung-Li, Taiwan, Republic of China
bNanotechnology & Center for Nanotechnology, Chung-Yuan Christian University, Chung-Li, Taiwan, Republic of China
First published on 7th October 2016
Abstract
Vinyl ester/inorganic layered material nanocomposites were prepared from osmotic swelled clay which is a typical inorganic layered material. Inorganic layered material (montmorillonite clay type (CL120), Zinc Aluminum Layer Double Hydroxide (ZnAl LDH)) based nanocomposites have dominated the polymer literature and is one of the most significant areas of current and emerging interest. Disodium cocoamphodipropionate (K2) and sodium dodecylbenzene sulfonate (SDBS) were intercalated into montmorillonite and ZnAl LDH, respectively. Modified CL120 and ZnAl LDH dispersion morphology is confirmed by using X-ray diffraction (XRD), thermogravimetric analysis (TGA) and Fourier transform infrared spectroscopy (FT-IR). The synthesized nanocomposites’ dispersion morphology is confirmed by XRD and TEM, and in order to evaluate the influence of these nanofillers, samples with different concentrations (1 and 3 phr) were dispersed in the matrix. The mechanical and shrinkage properties, dimensional stability, and print through phenomenon are expected to effectively help the development of wind blades.
1. Introduction
Since the last decade, wind power has been one of the promising renewable green sources that could replace fossil fuels and fulfill the rising global demand for sustainable energy. The demand for wind power and wind blades is increasing every year, thus calling for research into the environmental risks associated with the operation of large-scale commercial wind blades and ventures. Polymers and polymer based composite materials are used for the production of wind blades. These polymer composite materials are relatively low cost and are easily processed to improve the properties of wind blades.1,2Vinyl Esters (VEs) are frequently utilized in large-scale structural applications due to their low viscosity, ability to enable processing using resin infusion techniques, and ability to cure at ambient temperature. Usually, VE is made from the reaction of methacrylic or acrylic acids with an epoxy resin. VE polymers show excellent properties such as heat resistance and mechanical properties irrespective of the unsaturated polyester resin. VEs are used alone or in fiber-reinforced composites, in naval constructions, offshore applications, water pipes, chemical containers, building construction, and automotives, etc.3,4
Nanocomposites constitute one of the most recent areas of nanotechnology. Polymer/clay nanocomposites, specifically, have become increasingly popular in the past decade. Nanocomposites are extensively used as ideal materials to meet the challenge mentioned above, and the level of dispersion of these nanofillers throughout the polymer matrix assists with the enhancement of particular properties, such as the mechanical, thermal, flame retardant, optical, and gas barrier properties, etc.5–9 In this sense, layered double hydroxides (LDHs),10–16 montmorillonite,17–20 graphene and carbon nanotubes21–24 are being used in order to obtain these kinds of materials. So far, Gupta et al., Kalaee et al., Ratna et al., and Yah et al. have reported methods for preparing VE/clay nanocomposites. A composite prepared by high speed mechanical stirring followed by an ultra-sonic dispersion method, can improve its tensile strength, surface hardness, and the pyrolysis temperature. But, this method is not easy for mass production of nanocomposites, it needs a lot of energy and therefore development of a new method for mass production is desirable.
Fortunately, the osmotic swelling synthesis method can be altered for nanocomposite preparation. Basically natural clays and LDH nanolayers covalently bond to each other with interlayer ions bonded through van der Waals forces, ionic or hydrogen bonds. Interlayer exchangeable ions allow solvent molecule into layers get into swollen state laminate layers and increase the distance between the layers. In this paper, we describe the osmotic swelling method for in situ free radical polymerization of well dispersed VE nanocomposites. The dispersion morphology and mechanical properties of the obtained VE/clay nanocomposites were then investigated. Furthermore Barcol hardness, volume shrinkage and print-through phenomenon (PTP) results were discussed briefly.
2. Experimental
2.1 Materials
All the chemicals used were of analytical grade or of the highest purity commercially available. Montmorillonite (CL120, CEC 168 meq./100 g) with the molecular formula Ca0.16Na0.44(Al3.241Mg0.759)Si8O20(OH)4·0.37H2O was supplied by the China Glaze company, ZnAl-LDH (Zn3.832Al2.000(OH)11.663)(CO3)1.000·5.691H2O (AEC 300 meq./100 g) was provided by the Beijing University of Chemical Technology, and disodium cocoamphodipropionate (K2) was purchased from Lonza. Epoxy resin (Epoxy) NPEL-128 (Epoxy Equivalent Weight (EEW) of 186 to 190) was supplied by Nan Ya Plastics Corporation-Taiwan. Sodium dodecyl benzene sulfonate (SDBS), methacrylic acid (MAA), acetic acid and triphenylphosphine (TPP) were purchased from Sigma-Aldrich. Styrene Monomer (SM), Methyl Ethyl Ketone Peroxide (MEKPO), cobalt(II) naphthenate and hydroquinone (THQ) were purchased from Changxing industry Co. Ltd. and double distilled high-purity water was obtained from a Milli-Q water (Millipore) system.2.2 Preparation of organomodified clay (CL120-K2)
Experimentally, 5 g of CL120 was dispersed in 250 ml of deionized water and stirred at room temperature for 24 h to form swollen CL120. In this swollen CL120, K2 was added (1.5 times CEC of the CL120), and the pH of the resulting solution was maintained at 4–5 by adding 1 M acetic acid. This solution was rapidly agitated at 70–80 °C for 24 h. The obtained precipitate was collected by centrifugation and washed with deionized water, followed by freeze-drying. These synthesized samples are denoted CL120-K2.2.3 Preparation of organomodified ZnAl LDH
Commercial ZnAl LDH was calcined at 400 °C for 1 h to produce Zinc Aluminum Layered Double Oxide (ZnAl LDO). After that ZnAl LDO was reconstructed and modified with SDBS. SDBS (2 times the AEC of ZnAl LDH) dissolved in 250 ml of water to form a modifier aqueous solution. In this solution 5 g of ZnAl LDO was added slowly, and the resulting solution was placed in a high-pressure autoclave to react at 160 °C for 6 h to form a SDBS modified ZnAl LDH precipitate. The precipitate was collected by centrifugation and then washed with deionized water, followed by freeze-drying. These modified samples are denoted ZnAl–SDBS-LDH.2.4 Preparation of VE nanocomposites
Different concentrations of the organo-modified inorganic layered materials (CL120-K2, ZnAl–SDBS-LDH) were blended with 27.9 g of SM at room temperature for 24 h to swell the inorganic layered materials completely. 23.25 g of epoxy, 10.75 g of MAA and 0.010 g of THQ were charged into a round bottom flask and the mixture was stirred at 90 °C for 30 min. After that 0.1163 g of TPP was added, and the resulting reaction mixture was kept at 100 °C for 3 h. During the reaction, the acid value was maintained at ∼10. This solution was added to the above prepared swollen inorganic layered material solution. To this, a blended styrene solution of 0.3 phr cobalt(II) naphthenate and 1.2 phr MEKPO was added slowly under stirring. The resulting polymer solution was molded and cured at 40 °C for 4 h. Finally, the synthesized composites are represented as VE/CL120-K2-1.0 phr, VE/CL120-K2-2.0 phr, VE/CL120-K2-3.0 phr, VE/ZnAl-SDBS-LDH-1.0 phr, VE/ZnAl-SDBS-LDH-2.0 phr and VE/ZnAl-SDBS-LDH-3.0 phr.3. Characterization
X-ray diffraction was carried out on a PANalytical PW3040/60 X’Pert Pro, with Cu Kα radiation (45 kV, 40 mA) and wavelength at λ = 1.54 Å. The scan angle covered 2° < 2θ < 80° for layered materials and nanocomposites at a scan speed of 3 min−1. IR spectra were recorded with a JASCO, FTIR-4200 Type A spectrometer using KBr pellets. The morphology of VE nanocomposites was measured by scanning electron micrographs (SEM), and was obtained on a JEOL JSM 6500F. Transmission electron microscopy (TEM) images were obtained on a Model JEOL JEM2010, 200 kV. A sample with a thickness of 80 nm was prepared using a Leica Ultracut-UCT to verify the physicochemical properties of the layered materials. Thermogravimetric analysis (TGA) was carried out using SII TG/DTA6200, and the experiment was performed using a 10 mg sample of VE nanocomposites under air gas flow from 40 °C to 900 °C at a scanning rate of 10 °C min−1. The dynamic mechanical analysis (DMA) measurement was performed with a TA-Q800 instrument in the air at a scanning range of 30–210 °C, a heating rate of 5 °C min−1 and a sample size of 35 mm × 12 mm × 3 mm. Surface hardness was measured by using a Precisa/XS225A instrument. The tensile strength of the nanocomposites was measured by using Hongda instruments HT-9102M, where samples were prepared according to the ASTM D638 and D3039 specification and with a stretching rate of 1 mm min−1. Elemental analysis was carried out using Inductively Coupled Plasma Atomic Emission Spectroscopy, (ICP-OES) and Elemental Analysis (EA).4. Results and discussion
The pure CL120 and CL120-K2 diffraction patterns are shown in Fig. 1(a). In Fig. 1(a), the characteristic peak of CL120 is observed at 2θ = 6.94° (001) with a corresponding d-spacing of 12.74 Å, but after modification the peak lowers to 2θ = 2.10° (001) with a corresponding d-spacing of 40.01 Å. The d-spacing increases by about 27.24 Å, which is almost equal to the theoretical size of K2, 22.3 Å, providing evidence that the modifier K2 is intercalated into the galleries of inorganic layers.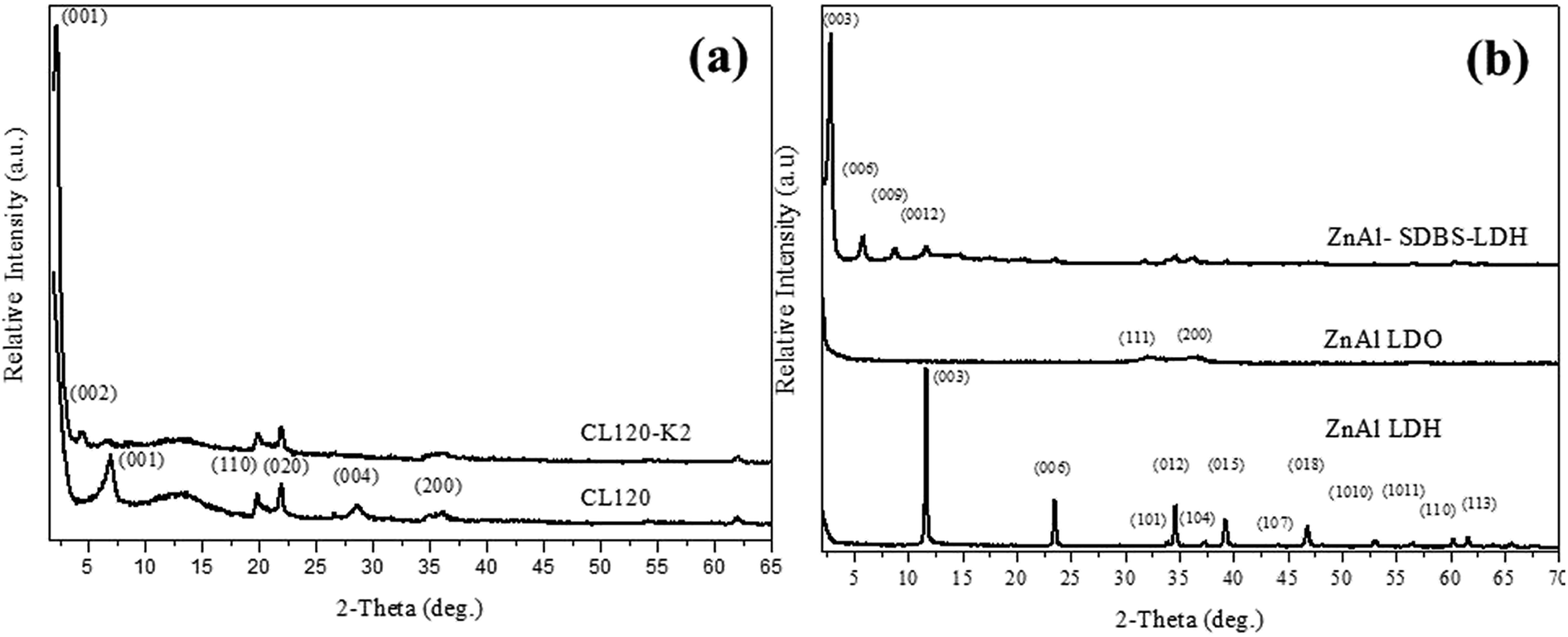 | ||
| Fig. 1 XRD patterns of (a) clay and modified clay, and (b) ZnAl LDH and ZnAl–SDBS-LDH. | ||
Synthesized ZnAl LDH and ZnAl–SDBS-LDH were characterized by using XRD. The pure ZnAl LDH’s highest order of reflection was obtained at 2θ = 11.4° (003) with a corresponding d spacing of 7.7 Å, and after modification (ZnAl–SDBS-LDH) the 2θ value was lowered and the basal spacing of the (003) phase increased up to 22.04 Å. The most drastic changes in the ZnAl LDH structure are due to SDBS chemically bonding with the interlayer of ZnAl LDH. The changes affecting the interlayer distances and the basal spacing increase. This provides evidence that SDBS is intercalated into the galleries of LDHs, as shown in Fig. 1(b). The variation of the d-spacing is caused by the orientation of SDBS, as the theoretical molecular size is 20.6 Å, which is almost equal to the d spacing of ZnAl–SDBS-LDH. The stronger intensity of the peaks indicates a greater crystallinity of the sample. Furthermore, the Zn/Al ratio of ZnAl-LDH was determined using ICP elemental analysis results summarized in Table 1.
| Sample | Elements | |||||
|---|---|---|---|---|---|---|
| Zn | Al | C | H | N | Zn/Al | |
| ZnAl LDH | 37.01 | 7.97 | 2.43 | 3.91 | 48.69 | 4.64 |
The formation of the highly reactive modified layered materials was also confirmed by FTIR spectroscopy. Fig. 2(a) shows the FT-IR spectra of the before and after modification of CL120 in the region of 400–4000 cm−1. The intense broad band at 3500 cm−1 is due to the stretching vibration of hydroxyl groups from the brucite-like layers; the band at 3626 cm−1 corresponds to the vibrations of water molecules from the interlayer. The weakness of this band should be related to the small amount of water existing in the hydrophobic interlayer. The weak bands at 2924 and 2854 cm−1 are due to the antisymmetric and symmetric modes of the C–H bonds, in the methylene groups. The bands at 1644 cm−1 and 1554 cm−1 correspond to the vibrations of amide and –NH bonds, respectively. The Mg–O and Al–O bands are recorded at 470 cm−1, 520 cm−1, and 1035 cm−1. The 1106 cm−1 band corresponds to Si–O–Si stretching vibrations.
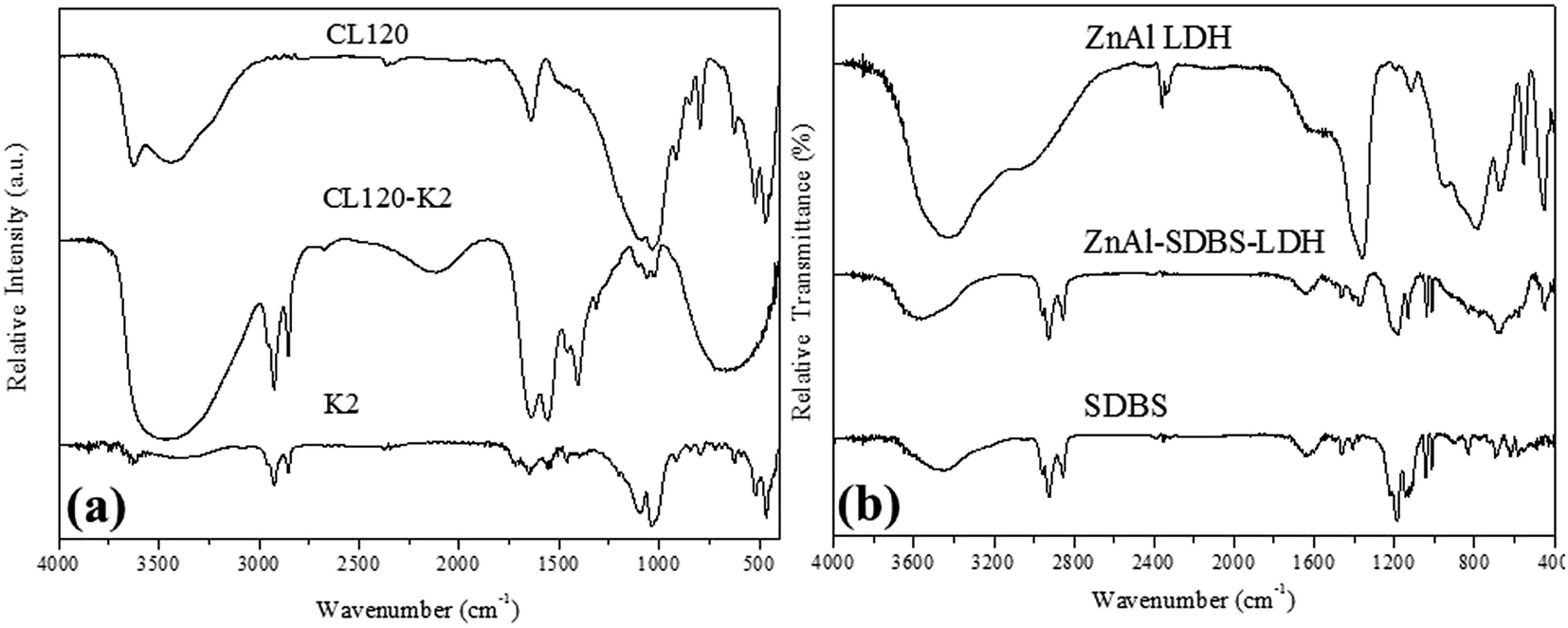 | ||
| Fig. 2 FT-IR spectra of (a) clay and modified clay, and (b) ZnAl LDH and ZnAl–SDBS-LDH. | ||
The FT-IR spectra of ZnAl LDH and ZnAl–SDBS-LDH are shown in Fig. 2(b), the bands at 2960, 2925 and 2852 cm−1 correspond to the stretching vibrations of the –CH bonds of the alkyl chain; the SDBS anion and other bands corresponding to the nitrate ion are recorded. Bands at 1183 and 1038 cm−1 represent the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric and symmetric stretching vibrations, respectively. 1131 and 1011 cm−1 bands correspond to the CH bending vibrations of the benzene ring.25 There are bands at low wavenumbers which correspond to the M–O, OMO, and MOM (M = Zn, Mg, Al) vibrations. This is evidence that SDBS is intercalated into the LDH layers.
O asymmetric and symmetric stretching vibrations, respectively. 1131 and 1011 cm−1 bands correspond to the CH bending vibrations of the benzene ring.25 There are bands at low wavenumbers which correspond to the M–O, OMO, and MOM (M = Zn, Mg, Al) vibrations. This is evidence that SDBS is intercalated into the LDH layers.
TGA curves of the layered materials are shown in Fig. 3(a) and (b). The thermal stability of the modified clay is greater than that of the pure clay. The plot for the pure clay exhibits a very small weight loss which commences at 120 °C, followed by maintained weight loss until 900 °C, but in the case of the modified clay, the weight loss started from 30–500 °C which represented the weight loss of surface adsorbed water, crystalline water and the –OH group of the inorganic layers. Pyrolysis of metal oxides occurred from 500–700 °C. ZnAl LDH and ZnAl–SDBS-LDH TGA curves are shown in Fig. 3(b), where weight loss is observed from 30–190 °C due to the loss of water and crystalline water, from 190–400 °C due to the loss of interlayer carbonates and hydroxide, and furthermore from 400–800 °C due to the loss of residual carbonate and the pyrolysis of metal oxide.
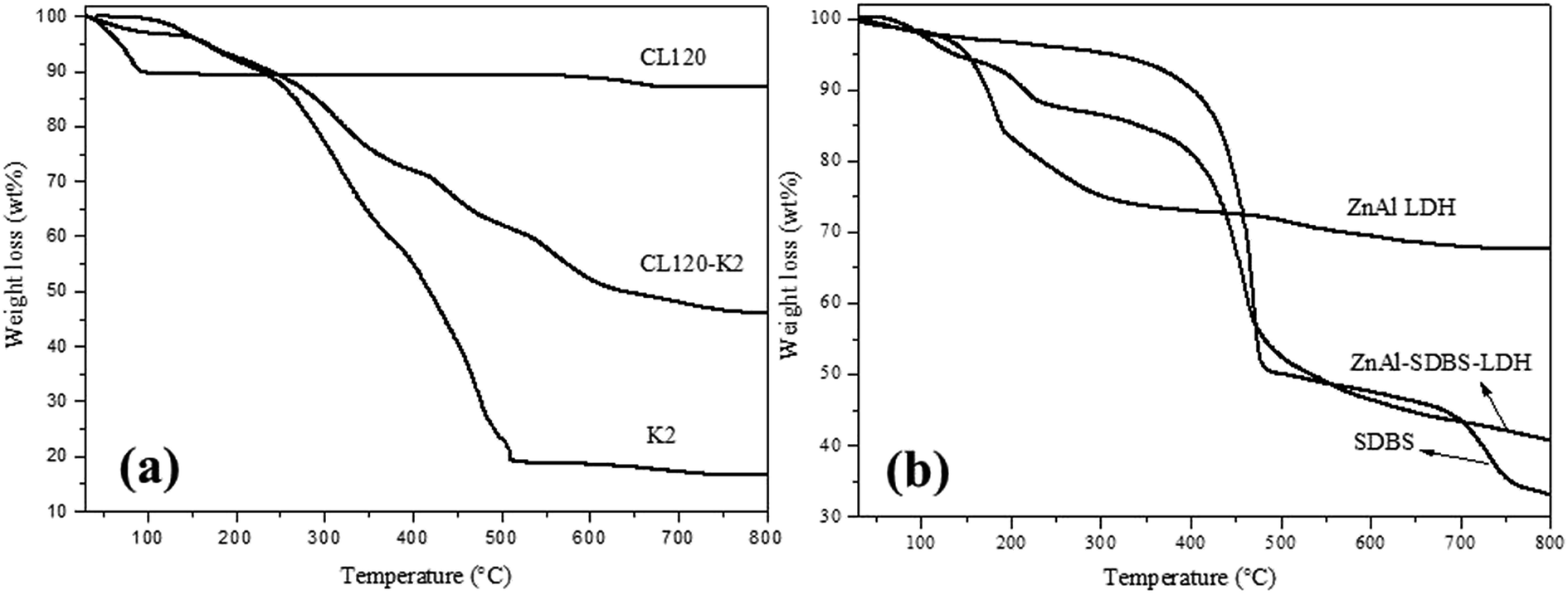 | ||
| Fig. 3 TGA curves of (a) pure clay and modified clay and (b) pure ZnAl LDH and modified ZnAl LDH. | ||
4.1 Morphology of VE nanocomposites
The VE nanocomposite morphology was confirmed by XRD. XRD patterns of the VE nanocomposites are shown in Fig. 4(a) and (b). The characteristic peak of CL120 completely disappeared, indicating that the nanolayers are well dispersed (intercalated or exfoliated) in the polymer matrix. In the case of the VE–ZnAl–SDBS-LDH nanocomposite 2.0 phr and 3.0 phr samples, the sharp peak shown at lower 2θ is due to the introduction of ZnAl–SDBS-LDH in the VE polymer matrix. This agreement provides strong evidence that the LDH is intercalated into the VE polymer matrix.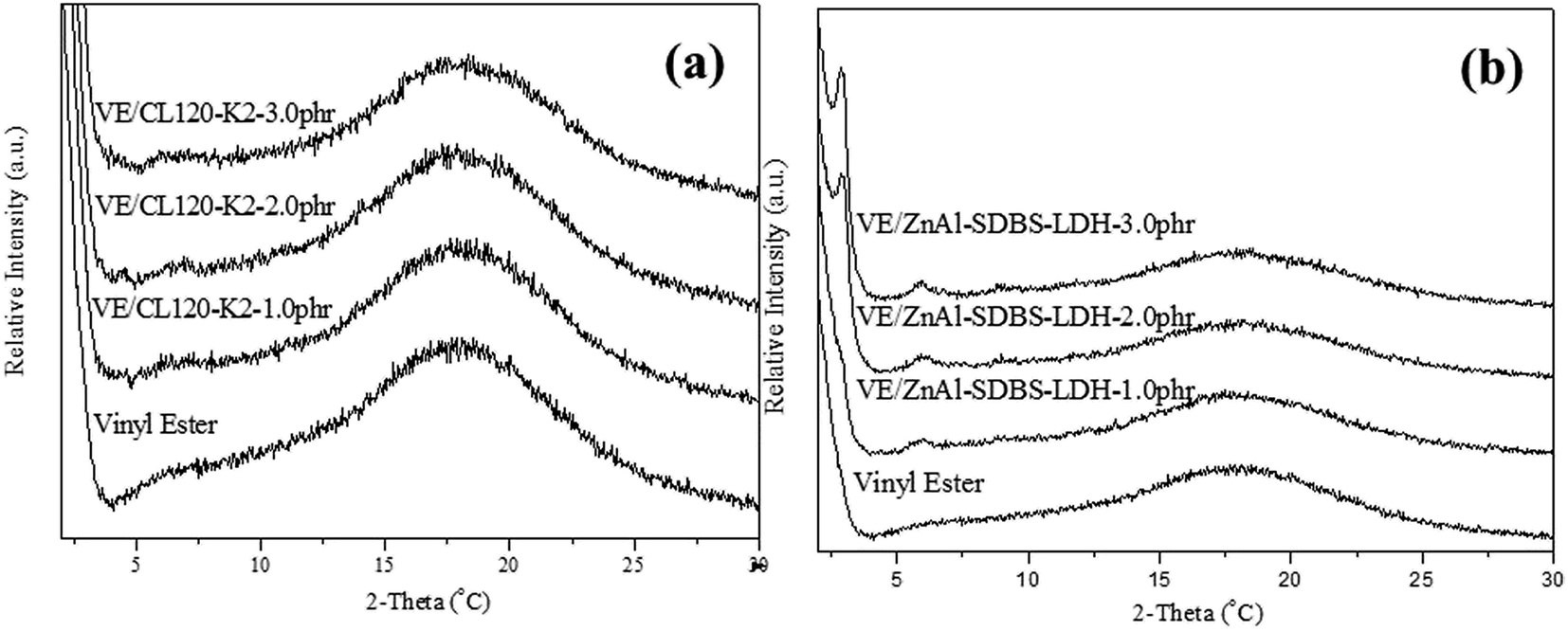 | ||
| Fig. 4 XRD patterns of the nanocomposites: (a) VE/CL120-K2 nanocomposites and (b) VE/ZnAl-SDBS-LDH nanocompistes. | ||
Nanocomposite dispersion morphology was further confirmed by the TEM analysis. As illustrated in Fig. 5(a) and (b) CL120-K2 nanosheets are delaminated and the island type of the intercalated dispersion is observed. The TEM images of VE–ZnAl–SDBS-LDH (Fig. 5(al) and (bl)) show all possible platelet morphologies, namely exfoliated, intercalated and stacked structures within the polymer matrix. It must be noted that a significant expansion of the LDH platelets has been observed even with only 2.0 phr and 3.0 phr mixing when in situ polymerization is carried out. This suggests that the monomer wets the LDH surface easily, and in the present case, there is very good compatibility between the surface treated SDBS and the vinyl ester resin.
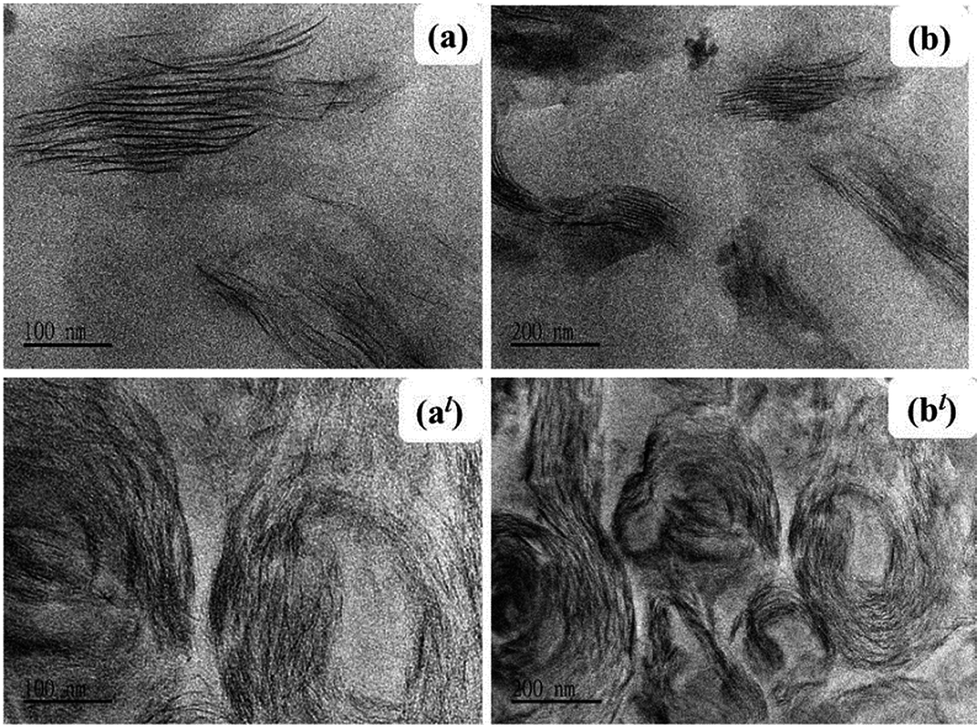 | ||
| Fig. 5 TEM images of (a) and (b) the VE/CL120-K2-2 phr nanocomposite, and (al) and (bl) the VE/ZnAl-SDBS-LDH-2 phr nanocomposite. | ||
4.2 Mechanical properties of VE nanocomposites
The storage modulus, glass transition temperature (Tg) and tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ curves for the pure VE and VE nanocomposites determined by DMA analysis, are all summarized in Table 2 and Fig. 6(a) and (b). It is observed that the VE nanocomposite Tg value is higher than that of the pure VE. However, increasing the concentration of the nanofillers leads to a decrease in the crosslink density and Tg. The crosslinking density will affect the polymer chain growth, so we controlled the amount of layered materials loading. We also observed that when we add a small amount of the nanofiller, it interferes with the flow of the polymer chain, hence reducing the crosslink density, and resulting in a lower Tg.
δ curves for the pure VE and VE nanocomposites determined by DMA analysis, are all summarized in Table 2 and Fig. 6(a) and (b). It is observed that the VE nanocomposite Tg value is higher than that of the pure VE. However, increasing the concentration of the nanofillers leads to a decrease in the crosslink density and Tg. The crosslinking density will affect the polymer chain growth, so we controlled the amount of layered materials loading. We also observed that when we add a small amount of the nanofiller, it interferes with the flow of the polymer chain, hence reducing the crosslink density, and resulting in a lower Tg.
| Sample Name | T5d (°C) | Tg (°C) | Tensile strength (MPa) | Elongation (%) | Young modulus | Hardness |
|---|---|---|---|---|---|---|
| Vinyl ester | 361 | 148.5 | 35.73 | 3.30 | 76.81 | 65 |
| VE/CL120-K2-1.0 phr | 358 | 168.5 | 42.39 | 4.04 | 79.31 | 69 |
| VE/CL120-K2-2.0 phr | 357 | 165.7 | 45.00 | 4.40 | 79.83 | 70 |
| VE/CL120-K2-3.0 phr | 356 | 160.5 | 51.71 | 5.20 | 81.50 | 70 |
| VE/ZnAl–SDBS-LDH-1.0 phr | 354 | 169.1 | 40.95 | 4.72 | 76.41 | 66 |
| VE/ZnAl–SDBS-LDH-2.0 phr | 348 | 166.3 | 56.37 | 5.55 | 79.51 | 67 |
| VE/ZnAl–SDBS-LDH-3.0 phr | 341 | 162.1 | 48.55 | 4.84 | 78.58 | 67 |
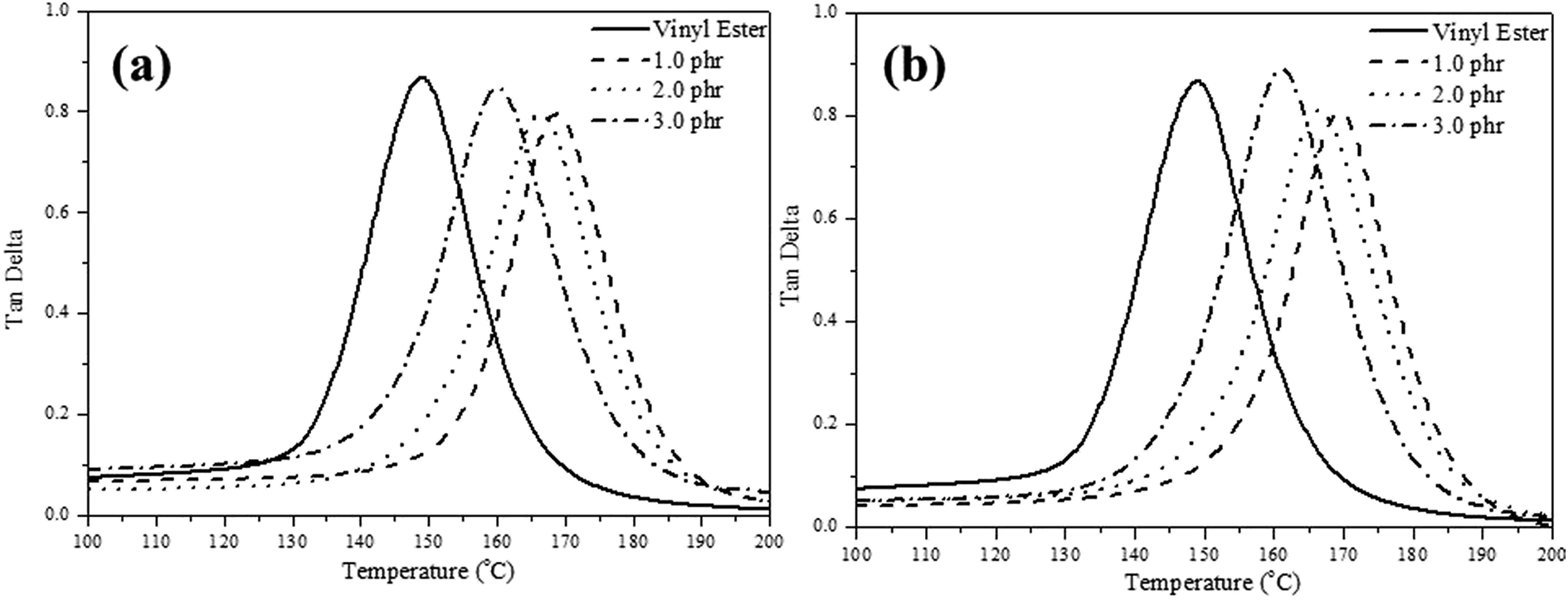 | ||
| Fig. 6 DMA temperature vs. tanδ curves of (a) VE/CL120-K2, and (b) VE/ZnAl–SDBS-LDH nanocomposites. | ||
Table 2 also presents the mechanical properties of the pristine VE and the VE nanocomposites with different nanofiller amounts. The tensile elongation and Young’s modulus of the nanocomposites are higher than that of the pristine VE. But in the case of the VE–ZnAl–SDBS-LDH nanocomposite 2.0 phr, the tensile modulus and elongation are higher than that of the 3.0 phr loaded nanocomposite, which might be due to better dispersibility, as it may be that the large portion of agglomeration in the 3.0 phr nanocomposite causes the decrease in the tensile module and elongation. FRP tensile test results are shown in Fig. 7(a), where the tensile strength of the VE-CL120-K2 and VE–ZnAl–SDBS-LDH nanocomposites increased from 338 MPa to 395 MPa and 384 MPa, respectively as compared to the pure VE. The tensile modulus increases due to the good dispersion of organomodified clay in the polymer matrix. ZnAl–SDBS-LDH loaded nanocomposites show a smaller improvement in the tensile test. This occurs due to the dispersion of the nanofiller in the VE matrix.
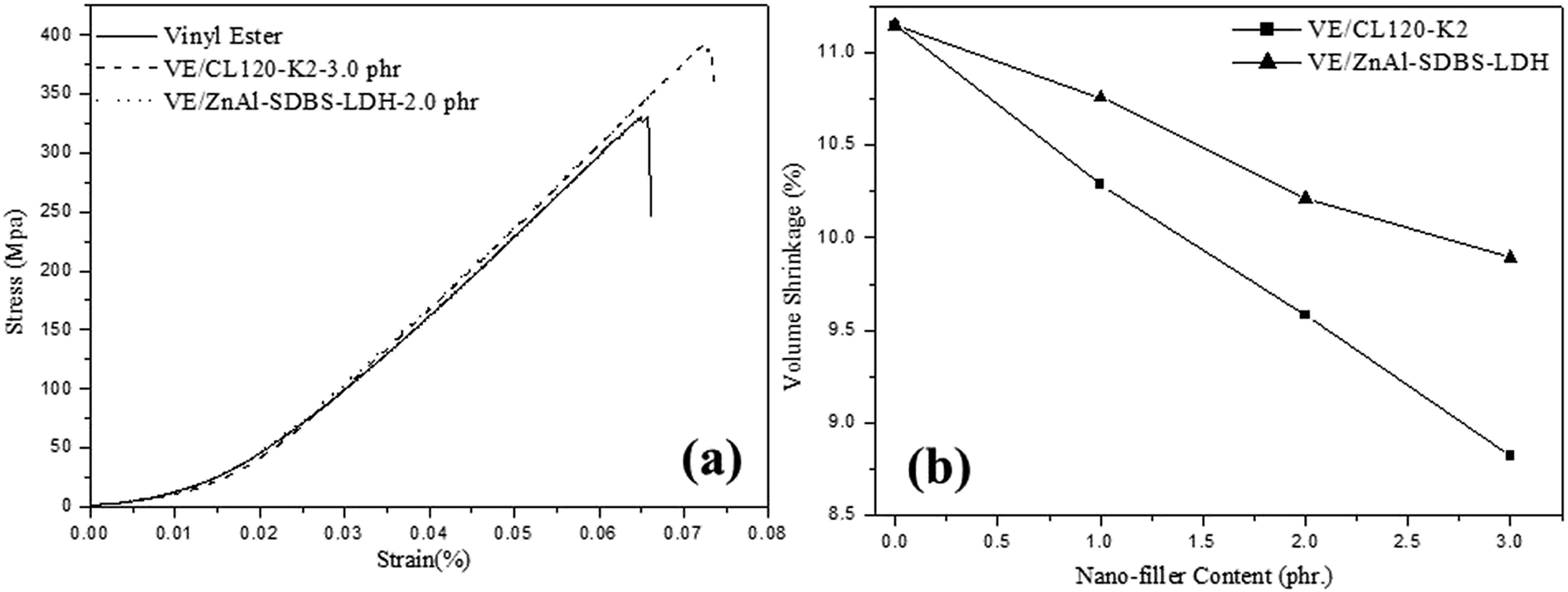 | ||
| Fig. 7 (a) Tensile strength test analysis, and (b) shrinkage properties of the VE nanocomposites. | ||
The results for the hardness properties of the pristine VE and VE nanocomposites are summarized in Table 2. The hardness of the VE nanocomposites increases with the incorporation of the nanofiller. All of the nanocomposites exhibit increased hardness as compared to that of the pure VE. The increase in the hardness is due to the presence of nanofillers in the polymer matrix. The nanofiller adequately restricts indentation and thus enhances the hardness of the nanocomposites. It is necessary to mention that the improvement of the hardness of the VE/CL120-K2 composites is significantly higher than that of the VE/ZnAl–SDBS-LDH nanocomposites. This is due to a large portion of agglomeration in the ZnAl–SDBS-LDH nanofiller in the polymer matrix.
4.3 Dimensional stability of VE nanocomposites
According to the results shown in Fig. 7(b) the shrinkage volume of the pure VE is higher compared to that of the VE nanocomposites. The nanocomposite shrinkage volume decreased by increasing the content of the nanofiller. This might be due to surface deformation of resin. It is also noticed that changing the curing temperature does not vary the resin conversion at the volume expansion point. This suggests that at different curing temperatures, the driving forces (i.e. polymerization shrinkage) at the expansion point are similar. Hence, the volume expansion observed at higher temperatures is probably due to lower solid modulus at higher cure temperatures, which provides less resistance to crack initiation and propagation of same resin with same morphology. Other factors, such as a higher shrinkage rate at higher curing temperatures may also contribute to microcracking at high temperatures.26 The shrinkage properties of VE/ZnAl–SDBS-LDH are higher than that of VE/CL120-K2, which is due to the fact that the general nature of the fracture surface morphologies of the two composites differ significantly.27A print-through phenomenon often occurs on the surface of the gelcoat layer of fiber-reinforced plastic (FRP). The fiber prints are visible with the naked eye and were investigated. Reflection images of fluorescent lamp on the surface of FRP specimen before curing and after curing are shown in Fig. 8. The images of the unsaturated VE nanocomposites were much clearer than those on the FRP specimens. Thus, the surface of the FRP specimen of the unsaturated VE/CL120-K2 nanocomposites was more even and smooth than that of the saturated FRP specimen. The print-through phenomenon was reduced. The different degrees of curing within the VE resin resulted in a differential thermal contraction of composites, therefore, a print-through phenomenon (PTP) appeared on the surface.28
 | ||
| Fig. 8 Comparison of the seal surface phenomena before and after curing. | ||
5. Conclusion
In the present study, the effect of the osmotic swelled nanofiller treatment on the in situ synthesis of VE nanocomposites has been studied. Nanocomposite morphology was studied by XRD and TEM analysis. Due to the osmotic swell of the nanofiller, VE/CL120-K2 nanocomposites show a homogenously dispersed morphology, whilst VE/ZnAl–SDBS-LDH shows an intercalated morphology. Besides the storage modulus, dimensional stability of the nanocomposites increased by increasing the nanofiller loading. Moreover by shaping FRP with a core-mat layer during manufacturing, the degree of the PTP problem can be reduced effectively. Overall, the use of the osmotic swelling method is effective in the dispersion of natural clay, which improves the mechanical properties and dimensional stability, and it is expected to effectively help in the development of wind blades.Acknowledgements
The authors are grateful to the Ministry of Science and Technology (MOST) Taiwan, the Ministry of Education, Taiwan, R. O. C. under Grant MOST103-2113-M-033-044 and MOST103-2622-M-033-001-CC2.References
- P. S. Apoorva, K. G. Rakesh, H. V. S. Gangarao and C. E. Powell, Polym. Eng. Sci., 2002, 42, 1799 Search PubMed.
- C. M. C. Pereira, M. Herrero, F. M. Labajos, A. T. Marques and V. Rives, Polym. Degrad. Stab., 2009, 94, 939 CrossRef CAS.
- S. Nazaré, B. K. Kandola and A. R. Horrocks, Polym. Adv. Technol., 2007, 17, 294 CrossRef.
- C. M. C. Pereira and A. T. Marques, in Proceedings of the 12th European conference on composite materials, Biarritz, France, September, 2006 Search PubMed.
- F. R. Costa, M. Saphiannikova, U. Wagenknecht and G. Heinrich, Adv. Polym. Sci., 2007, 210, 101 CrossRef.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS PubMed.
- J. L. Gunjakar, T. W. Kim, H. N. Kim, I. Y. Kim and S.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 14998 CrossRef CAS PubMed.
- J. Tronto, A. Cludia, Z. Naal and J. B. Valim, Conducting Polymers/Layered Double Hydroxides Intercalated Nanocomposites, INTECH, 2013 Search PubMed.
- J. Zhao, J. Chen, S. Xu, M. Shao, Q. Zhang, F. Wei, J. Ma, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2014, 24, 2938 CrossRef CAS.
- B. Guo, Y. Zhao, Q. Huang and Q. Jiao, Compos. Sci. Technol., 2013, 81, 37–41 CrossRef CAS.
- S. Xu, L. Zhang, Y. Lin, R. Li and F. Zhang, J. Phys. Chem. Solids, 2012, 73, 1514 CrossRef CAS.
- Y. Yuan and W. Shi, Prog. Org. Coat., 2010, 69, 92 CrossRef CAS.
- J. Yang, F. Chen, Y. Ye, Z. Fei and M. Zhong, Colloid Polym. Sci., 2010, 288, 761 CAS.
- C. M. C. Pereira, M. Herrero, F. M. Labajos, A. T. Marques and V. Rives, Polym. Degrad. Stab., 2009, 94, 939 CrossRef CAS.
- Z. Hu and G. Chen, Adv. Mater., 2014, 26, 5950 CrossRef CAS PubMed.
- Z. Hu and G. Chen, J. Mater. Chem. A, 2014, 2, 13593 CAS.
- M. Galimberti, Chem. Mater., 2007, 19, 2495 CrossRef CAS.
- S. Nazarenko, P. Meneghetti, P. Julmon, B. G. Olson and S. Qutubuddin, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1733 CrossRef CAS.
- J. P. Zheng, L. Luan, H. Y. Wang, L. F. Xi and K. D. Yao, Appl. Clay Sci., 2007, 36, 297 CrossRef CAS.
- A. A. Azeez, K. Y. Rheea, S. J. Park and D. Hui, Composites, Part B, 2013, 45, 308–320 CrossRef CAS.
- S. Y. Yang, C. C. M. Ma, C. C. Teng, Y. W. Huang, S. H. Liao, Y. L. Huang, H. W. Tien, T. M. Lee and K. C. Chiou, Carbon, 2010, 48, 592 CrossRef CAS.
- K. Yang, M. Gu, Y. Guo, X. Pan and G. Mu, Carbon, 2009, 47, 1723 CrossRef CAS.
- X. He, F. Zhang, R. Wang and W. Liu, Carbon, 2007, 45, 2559 CrossRef CAS.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- F. R. Costa, M. Abdel-Goad, U. Wagenknecht and G. Heinrich, Polymer, 2005, 46, 4447 CrossRef CAS.
- W. Li and L. J. Lee, Polymer, 2000, 41, 697 CrossRef CAS.
- A. Al-Khanbashi, M. El-Gamal and A. Moet, J. Appl. Polym. Sci., 2005, 98, 767 CrossRef CAS.
- S. R. White and H. T. Hahn, J. Compos. Mater., 1992, 26, 2402 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |