
Structural distortion and electronic states of Rb doped WO3 by X-ray absorption spectroscopy
 *ad
and
*ad
and
Abstract
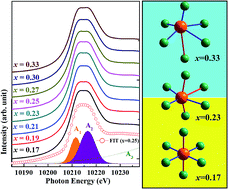
Rubidium tungsten bronzes (RbxWO3) have recently attracted much attention due to their intriguing phenomena, such as complex structural phase transitions, strong electron–phonon coupling, and superconducting properties. This study investigates the local atomic and electronic structures of RbxWO3 (0.17 ≤ x ≤ 0.33). X-ray powder diffraction patterns showed a hexagonal tungsten bronze (HTB) phase. X-ray absorption spectra (XAS) at the W L3-edge and Rb K-edge of RbxWO3 were carried out. The XAS analysis indicated a local distorted WO6 octahedron which leads to a splitting of eg and t2g energy states in the tungsten 5d orbital and this splitting of energy levels exhibited an asymmetrical behavior at x = 0.23 and 0.27. Overall analysis revealed a distortion of local atomic structure of the WO6 octahedra by rubidium doping, leading to the modification of the electronic structures of eg and t2g states in the tungsten 5d orbital, thereby accounting for the property changes in CDW formation and superconducting transition temperature of these materials.
 Please wait while we load your content...
Please wait while we load your content...

