DOI:
10.1039/C6RA21716H
(Paper)
RSC Adv., 2016,
6, 107141-107150
Electrochemical properties of silver nanoparticle-supported reduced graphene oxide in nitric oxide oxidation and detection†
Received
30th August 2016
, Accepted 3rd November 2016
First published on 4th November 2016
Abstract
This article reports the effect of ascorbic acid in the formation of a reduced graphene oxide–silver (rGO–Ag) nanocomposite and its influence on the electrochemical oxidation of nitric oxide (NO). The formation of the rGO–Ag nanocomposite was confirmed using UV-visible absorption spectroscopy, X-ray diffraction (XRD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM) analyses. Crystalline and spherical Ag nanoparticles (NPs) with an average particle size of 2 nm were found in the rGO–Ag nanocomposite with the assistance of 5.0 M ascorbic acid. The electrochemical properties of the rGO–Ag nanocomposite-modified glassy carbon (GC) electrode were investigated for the oxidation of NO. The rGO–Ag (5.0 M) nanocomposite-modified electrode displayed a higher catalytic current response compared to other controlled modified electrodes in cyclic voltammetry toward the oxidation of NO in 0.1 M phosphate buffer (pH 2.5). The electroanalytical application of the nanocomposite was performed using an amperometry technique, and the limit of detection (LOD) was found to be 2.84 μM for the in situ detection of NO. The present nanocomposite was stable, sensitive, and selective in the presence of the common physiological interferents such as ascorbic acid, uric acid, dopamine, glucose, urea, and NaCl.
1. Introduction
Nitric oxide (NO) is one of the intracellular molecules, playing roles in the immune system and vasodilation pathway of the human nervous system.1,2 An excessive deficiency of NO results in various pathological conditions, which may lead to atherosclerosis,3 diabetes,4 angiogenesis,5 Parkinson's disease, and Alzheimer's disease.6 At a certain physiological pH, the submicromolar concentration of NO is hard to determine because it is a quickly diffusible gas molecule with a short lifetime (∼5 s).7 This characteristic makes it a challenging and attractive task to develop a sensitive NO sensor. A variety of methods have been used to detect NO.8 The most common methods for NO detection are UV-visible spectroscopy,9 electron paramagnetic resonance,10 fluorescence,11 chemiluminescence12 and electrochemistry.13 Almost all of the spectroscopic approaches require specific chemical labels with a large volume of analyte, which brings a high cost, along with a limited NO detection ability in real time.14 An electrochemical sensor was selected as one of the most suitable methods for real time NO detection in biological media because this technique provides the lowest concentration of spatially resolved concentration data.15
Noble transition metal nanoparticles have usually been used as the electrode materials for catalytic applications.16,17 Among many noble metal nanoparticles, silver nanoparticles (AgNPs) are in high demand in sensor applications because of their exceptional properties, which include biocompatibility, low toxicity, and the ability to support electrocatalytic activity.18 Nevertheless, a strong van der Waals force between AgNPs has the tendency to cause severe aggregation in a solution, which results in a precipitous loss in the sensitivity of the electrochemical activity. Because AgNPs can be easily immobilized on several inorganic and organic support materials,19–21 it is interesting to further study a method to protect these metal NPs from agglomeration, as well as improve their stability and enhance their electrocatalytic activity.
Recently, graphene has been demonstrated to be an extraordinary host material for either metal or metal oxide NPs.22 Graphene is an ideal catalyst support to anchor metal nanoparticles and offers a sensitive and selective catalytic performance because of its large specific surface area (2630 m2 g−1), high electrical conductivity, and good thermal and mechanical properties.23 Nowadays, the in situ preparation of a graphene–silver nanocomposite has become one of the most notable areas of research, and has been shown to be very simple and effective.24,25 Among the recognized approaches, the chemical reduction method has been revealed to be one of the most promising methods for the one pot reduction of graphene and metal nanoparticles because of its facile synthetic nature, and the fact that it is a scalable, controllable, and reproducible technique.26 However, most of the reagents used to reduce metal ions, such as sodium borohydride and hydrazine, are also toxic and acute.27 Therefore, a facile and environmentally friendly method for fabricating a reduced graphene oxide–silver (rGO–Ag) nanocomposite is still needed.
In this study, ascorbic acid was chosen as a reducing agent because it is a naturally occurring organic compound with antioxidant properties. Ascorbic acid will donate two electrons from a double bond between the second and third carbons of the 6-carbon molecule. The identified physiological and biochemical activities of ascorbic acid are the result of its action as an electron donor, which allows it to play the role of a reducing agent to prevent other compounds from oxidation. We here report on the preparation of rGO–Ag nanocomposites at different concentrations of ascorbic acid via a facile one-pot synthesis method. These nanocomposites were then applied as a modified electrode for the electrochemical oxidation and sensing of NO. The nanocomposite-modified GC (GC/rGO–Ag) electrode showed a synergistic behavior between the rGO and Ag NPs in the electrocatalytic oxidation of NO. The rGO–Ag (5.0 M ascorbic acid) nanocomposite-modified electrode was able to detect NO with a detection limit of 2.84 μM using an amperometric i–t curve technique. The proposed sensor was stable, reproducible, and selective toward the detection of NO.
2. Experimental section
2.1. Materials
Graphite flakes were procured from Asbury Inc. (USA). Silver nitrate (AgNO3) (98%), potassium permanganate (KMnO4) (98%), and sodium nitrite (NaNO2) were bought from R&M Chemicals. L-Ascorbic acid (99%) was purchased from Sigma Aldrich. Monosodium phosphate (NaH2PO4), sulphuric acid (H2SO4, 98%), phosphoric acid (H3PO4), hydrochloric acid (HCl), and hydrogen peroxide (H2O2, 30%) were obtained from Systerm (http://www.haiousaintifik.com). All of chemicals were of analytical grade and used as received without further refinement. Double distilled water was used to prepare the solutions for all the experiments.
2.2. Synthesis of rGO–Ag nanocomposite
GO was synthesized by following the simplified Hummer's method.28 For a simple and inexpensive chemical synthesis, the rGO–Ag nanocomposite was prepared by directly reducing both silver ions and GO using ascorbic acid as a reducing and stabilizing agent. The rGO–Ag nanocomposite was synthesized as follows. First, 0.5 M/22.02 g of ascorbic acid was dissolved in 15 mL of GO solution (1.0 mg mL−1) and stirred for 15 min. Then, 10 mL of a [Ag(NH3)2]+ complex containing 0.06 M AgNO3 and 0.5 mol L−1 ammonia was added to that solution and stirred for 6 h. The same method was adopted to prepare the nanocomposite with different concentrations of ascorbic acid (1.0 M and 5.0 M). After the stirring was completed, the mix was set aside to sit undisturbed at room temperature for 2 h. The color of the GO changed from brown to muddy green, which confirmed the formation of the rGO–Ag nanocomposite. The slurry-like product was centrifuged at 4000 rpm and washed five times with distilled water to remove the impurities. The final product was re-dispersed in 25 mL of distilled water and used for further analyses. The rGO–Ag nanocomposites prepared with 0.5 M, 1.0 M, and 5.0 M concentrations of ascorbic acid are represented as rGO–Ag (0.5 M), rGO–Ag (1.0 M), and rGO–Ag (5.0 M), respectively.
2.3. Electrochemical studies
All the electrochemical experiments were carried out in a three-electrode electrochemical cell system at room temperature. The modified glassy carbon (GC) electrode and platinum wire were used as the working and counter electrodes, respectively. A silver/silver chloride (Ag/AgCl) electrode was applied as a reference electrode. Prior to the modification, the GC electrode was polished with alumina slurry (0.05 μm) and cleaned by potential cycling between +1 and −1 V in 0.1 M H2SO4. The rGO–Ag nanocomposite-modified GC electrode (GC/rGO–Ag) was fabricated by drop-casting 5 μL of an aqueous rGO–Ag solution onto a GC electrode (d = 3 mm) surface and allowing it to dry at room temperature (25 °C) for 2 h. This rGO–Ag-modified electrode was used for the electrochemical oxidation and sensing of NO. Phosphate buffer (pH 2.5) was employed as the supporting electrolyte, and NaNO2 was used as the precursor to generate the NO in solution. For real sample detection, the real water samples were filtered to remove the particulate materials prior to the analysis. Different amounts of NO were added to the real water samples and the concentration of NO in the spiked water samples was detected using the proposed rGO–Ag nanocomposite sensor.
2.4. Characterization techniques
The UV-visible absorption spectrum of the GO–Ag nanocomposite was recorded on a Thermo scientific Evolution 300 instrument. The Raman spectra were evaluated using a Renishaw inVia Raman microscope with green laser excitation (the 532 nm line of a He–Ne laser as the excitation source). X-ray photoemission spectroscopy (XPS) was performed at the photoemission spectroscopy (PES) beamline, BL3.2a, of the Synchrotron Light Research Institute in Thailand to examine the bonding of the samples. The X-ray diffraction (XRD) pattern was determined with a Siemens D5000 instrument using copper Kα radiation (λ = 1.5418 Å) at a scan rate of 0.02° s−1. A transmission electron microscopy (TEM) analysis was performed with a JEOL JEM-2100F instrument operated at 200 kV. Electrochemical characterization was carried out using a PAR-VersaSTAT-3 Electrochemical and Metrohm Autolab Nova 1.11 Workstation.
3. Results and discussion
3.1. Characterization of rGO–Ag nanocomposite
The UV-visible spectra were obtained in an attempt to confirm the formation of the Ag nanoparticles on rGO sheets. The absorption spectra of the rGO–Ag nanocomposites prepared at different concentrations of ascorbic acid (0.5 M, 1.0 M, and 5.0 M) showed a characteristic of the surface plasmon resonance (SPR) band of Ag NPs at wavelengths of 433, 425, and 405 nm, respectively. As is well known, a GO solution displays a shoulder peak at around 300 nm and a maximum absorption peak at 228 nm (Fig. 1 (inset)), which are attributed to the π–π* transition of the atomic C–C bonds and n–π* transitions of the C–O group, respectively.29 The rGO–Ag (0.5 M) nanocomposite showed a broad SPR band for the Ag NPs, which verified the formation of larger spherical Ag NPs (Fig. 1a). The appearance of a broad absorption spectrum may have been because of the aggregation formation or poly-dispersion of the Ag NPs when using a smaller amount of ascorbic acid as a reducing agent. For the rGO–Ag (1.0 M) nanocomposite, the Ag NPs spectrum presented a narrower band with a very sharp peak (Fig. 1b), whereas the rGO–Ag (5.0 M) nanocomposite showed an intense SPR band for the Ag NPs centered at 403 nm (Fig. 1c), which is attributed to the formation of mono-dispersed spherical Ag NPs with a particle size smaller than that obtained in low concentrations of ascorbic acid (0.5 M and 1.0 M). The pattern of the SPR band and color of the rGO–Ag nanocomposite were influenced by their shape, size, and surroundings.30 A visible color variation in the rGO–Ag (0.5 M, 1.0 M, and 5.0 M) nanocomposites indicated a change in the aggregation state or size and shape of the Ag NPs.31 As is well known, the ascorbate ion is a weak reducing agent with a low antioxidation ability. It will undergo the oxidation process by the loss of one electron to become a radical cation and then lose a second electron to form dehydroascorbic acid. Since ascorbic acid is a natural antioxidant that is essential for numerous metabolic functions in living organisms, dehydroascorbic acid will naturally react with oxidants of the reactive oxygen species in ascorbic acid and form stable nanosized Ag particles.32 In addition, the formation of the [Ag(NH3)2]+ complex protected the fast reduction of Ag+ ions, which controlled the growth of the Ag NPs. The oxygen functionalities of the GO provided a stable support for the controlled formation of Ag NPs.
 |
| | Fig. 1 UV-visible absorption spectra obtained for rGO–Ag nanocomposite solutions (a: 0.5 M, b: 1.0 M and c: 5.0 M). Inset shows a UV-visible absorption spectrum of GO solution. | |
A TEM analysis was used to study the morphology and particle size of the Ag NPs in the rGO–Ag nanocomposite. Representative TEM images are shown in Fig. 2 to provide a clearer understanding of the morphologies of the rGO–Ag nanocomposites prepared at different concentrations of ascorbic acid. The TEM image of GO shows the formation of wide and flat sheets because of the disruption in the van der Waals interactions between the GO layers that occurred during the sonication process (Fig. 2A). The TEM image of rGO–Ag (0.5 M) shows the formation of fewer large particles with a high dispersion of smaller Ag seed particles (Fig. 2B and C). This image corroborates the broad absorption of rGO–Ag (0.5 M) in the UV-vis spectrum. The average size of the Ag NPs was controlled to 15 nm with 1.0 M ascorbic acid. However, the population of Ag NPs in the GO matrix was still low, as shown in Fig. 2D and E. A high population of Ag NPs with an average particle size of 2 nm was observed when using 5.0 M ascorbic acid, as displayed in Fig. 2F and G. This reveals the efficient formation of spherical Ag NPs, and the image agrees with the relatively sharp absorption peak for Ag NPs in the SPR spectra. In addition, the rGO present in the rGO–Ag nanocomposite shows irregular and layer structured sheets, which suggests the reduction of GO during the synthesis (Fig. 2F). For the rGO–Ag (0.5 M, 1.0 M, and 5.0 M) nanocomposites, the d spacing values measured from the lattice fringes of high-resolution images are 2.604, 2.083, and 1.563 Å, which are close to the d spacing values corresponding to the (1 1 1), (2 0 0), and (2 2 0) crystal planes of Ag, respectively (JCPDS no. 89-3722).33
 |
| | Fig. 2 TEM images for GO (A) and different magnifications of rGO–Ag nanocomposite prepared with different concentration of ascorbic acid (B & C: 0.5 M, D & E: 1.0 M, F & G: 5.0 M). | |
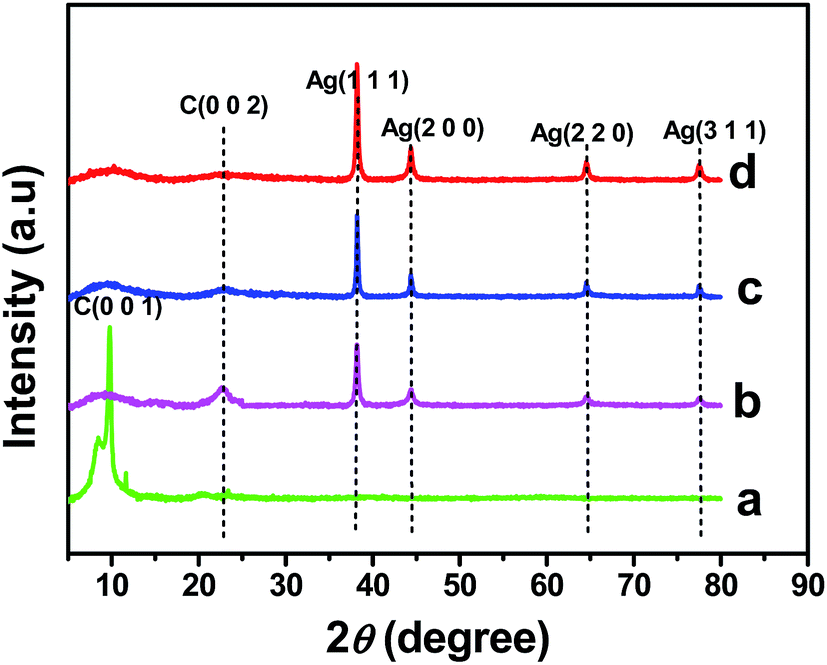
The formation and crystalline nature of the Ag NPs present in the rGO–Ag nanocomposites prepared at different concentrations of ascorbic acid were confirmed using XRD analysis. The XRD pattern of GO (Fig. 3a) shows a typical diffraction peak of C (0 0 1) at 10.5°.34 Fig. 3b–d shows the XRD patterns of the rGO–Ag (0.5 M, 1.0 M, and 5.0 M) nanocomposites. The four conspicuous peaks that appear at 38.2°, 44.3°, 64.4°, and 77.4° were indexed to the (1 1 1), (2 0 0), (2 2 0), and (3 1 1) crystallographic planes of the face centered cubic (fcc) of Ag particles, respectively (JCPDS no. 89-3722).35 The intensity of the Ag diffraction peaks increased with an increase in the ascorbic acid concentration (0.5 M, 1.0 M, and 5.0 M) in the rGO–Ag nanocomposite synthesis, while the peak intensity of the GO was concealed during the formation of the rGO–Ag nanocomposite and replaced by a new diffraction peak that appeared at 23.3°, which indicated the reduction of GO. Based on the rGO–Ag (0.5 M, 1.0 M, and 5.0 M) nanocomposites shown in Fig. 3b–d, the broad diffraction peak at 23.3° suggests that GO was reduced to rGO in the presence of different concentrations of ascorbic acid. However, the peak at 23.3° disappears in the XRD patterns of the rGO–Ag nanocomposite prepared with 5.0 M ascorbic acid, suggesting that the regular layered structure of the rGO was completely exfoliated as the amount of reducing agent increased. This was attributed to the insertion and uniform distribution of small AgNPs into the rGO sheets, which prevented the restacking of the layered structure of the rGO.36
 |
| | Fig. 3 XRD patterns of GO (a) and rGO–Ag nanocomposite prepared at different concentration of ascorbic acid (b: 0.5 M, c: 1.0 M and d: 5.0 M). | |
An XPS analysis was performed to verify the reduction of the GO during the chemical reaction process using ascorbic acid as a reducing agent (Fig. 4). For the GO sample, the XPS spectrum showed three main types of C 1s peaks at 284.7 eV, 286.8 eV, and 288.3 eV, which were allocated to C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.37 As shown in Fig. 4A–C, the C1 band of the rGO–Ag nanocomposites showed that the peak intensity of the oxygenated carbonaceous bands gradually decreased when the concentration of reducing agent was increased. The appearance of the significant Ag 3d5/2 and Ag 3d3/2 peaks at 368.7 and 372.3 eV, respectively, for metallic Ag confirmed the successful formation of Ag particles, together with the reduction of GO (Fig. 4D).38
O, respectively.37 As shown in Fig. 4A–C, the C1 band of the rGO–Ag nanocomposites showed that the peak intensity of the oxygenated carbonaceous bands gradually decreased when the concentration of reducing agent was increased. The appearance of the significant Ag 3d5/2 and Ag 3d3/2 peaks at 368.7 and 372.3 eV, respectively, for metallic Ag confirmed the successful formation of Ag particles, together with the reduction of GO (Fig. 4D).38
 |
| | Fig. 4 XPS spectra of rGO–Ag nanocomposites prepared at different concentration of ascorbic acid (A: 0.5 M, B: 1.0 M and C: 5.0 M) and XPS peaks of Ag (D). | |
The reduction of GO was doubly confirmed by the typical peaks indicated in a Raman spectral analysis (Fig. 5). The Raman spectrum of GO displayed the D and G bands at around 1350 cm−1 and 1600 cm−1, which correspond to the breathing mode of k-point phonons of A1g symmetry and the E2g phonon of C sp2 atoms, respectively (Fig. 5a).39 The intensity ratio between the D and G bands (ID/IG) is associated with the sum of disorder or the size of the sp2 domains.40 Referring to Fig. 5a–d, the value of ID/IG for GO is 0.96, while the intensity ratios (ID/IG) for the rGO–Ag nanocomposites prepared at 0.5 M, 1.0 M, and 5.0 M are 1.00, 1.01, and 1.03, respectively. The ID/IG intensity increased with the amount of reducing agent in the nanocomposite, which suggested the reduction of GO to rGO and is consistent with the XPS results.
 |
| | Fig. 5 Raman spectra of GO (a) and rGO–Ag nanocomposite with different concentration of ascorbic acid (b: 0.5 M, c: 1.0 M and d: 5.0 M). | |
3.2. Electrochemical behavior of rGO–Ag nanocomposite-modified electrode
The redox behavior of the [Fe(CN)6]3−/4− couple was used to study the kinetic barrier of the modified electrode solution interface. As recognized, the bare GCE showed the reversible voltammetric characteristic of the one electron redox process of the [Fe(CN)6]3−/4− couple with a peak-to-peak separation of 58 mV (Fig. 6A(a)). After modification with GO, the GCE lost its reversible voltammetric behavior because of the non-conducting nature of the GO matrix (Fig. 6A(b)). However, the reversibility was retained after modification with the rGO–Ag nanocomposite because of the high conductivity of Ag and the presence of rGO (Fig. 6A(c–e)). The rGO–Ag (5.0 M) nanocomposite showed higher redox peak currents compared to the rGO–Ag (0.5 M and 1 M) nanocomposites. This observation indicated that the rGO–Ag (5.0 M) nanocomposite acted as a new surface on the GCE and exhibited good electrical contact with the underlying electrode surface.
 |
| | Fig. 6 (A) Cyclic voltammograms obtained for bare GCE (a), GO (b), rGO–Ag (0.5 M) (c), rGO–Ag (1.0 M) (d) and rGO–Ag (5.0 M) (e) nanocomposites for 1 mM K3[Fe(CN)6] in 0.1 M KCl at a scan rate of 50 mV s−1. (B) Nyquist plot obtained for rGO–Ag (5.0 M) nanocomposite for 1 mM K3[Fe(CN)6] in 0.1 M KCl and the corresponding equivalent circuit diagram. Inset shows the Nyquist plots obtained for rGO–Ag (0.5 M) (a) and rGO–Ag (1.0 M) (b) nanocomposites. | |
The Nyquist plot observed for bare GCE showed a large semicircle that acted as a barrier for the electron-transfer kinetics at the electrode surface (Fig. S1(a)†). As evidenced from Fig. 6B, the Rct of the GCE electrode was enormously decreased by the modification with the rGO–Ag (0.5 M, 1.0 M, and 5.0 M) nanocomposite. The electron transfer was highly limited at the GO-modified electrode surface (Fig. S1(b)†). The rGO–Ag facilitated the interfacial electron transfer kinetics at the modified electrode–electrolyte solution interface because of the higher conductivity exhibited by the nanocomposite. The rGO–Ag (5.0 M) nanocomposite showed a perfectly linear portion at low frequencies compared to the other two nanocomposite-modified electrodes. These results confirmed that the rGO–Ag (5.0 M) nanocomposite was successfully coated on the electrode surface and controlled by a diffusion-limited process at the electrode–solution interface. The Nyquist plot obtained for the rGO–Ag (5.0 M) nanocomposite was fitted using an equivalent circuit model with ZSimpWin software (Fig. 6B (inset)). The equivalent circuit was comprised of several parameters, including the solution resistance Rs, which is in series with the double layer capacitance Cdl in parallel with Rct. The Warburg impedance (Zw) resulted from the diffusion of the redox analyte, whereas the interfacial properties of the nanocomposite were represented by Rct. The impedance values obtained from the fitted impedance spectrum are provided in Table S1.† The details of Bode phase and Bode impedance plots are provided in ESI (Fig. S2†).
3.3. Electrocatalytic oxidation of NO
An evidence of the presence of Ag at the rGO–Ag nanocomposite modified electrode and the coverage of Ag NPs on GCE surface are discussed in ESI (Fig. S3 and S4†). The electrocatalytic activity of the rGO–Ag nanocomposite-modified electrode was systematically investigated in relation to the oxidation of NO in the presence of 0.1 M phosphate buffer (pH 2.5). NaNO2 was selected as the precursor for the in situ production of NO in phosphate buffer. NO could be generated from NaNO2 in an acidic solution with a pH of less than four following the disproportionation reaction (eqn (1)).41,42| | |
3HONO → H+ + 2NO + NO3− + H2O
| (1) |
Fig. 7 shows the cyclic voltammetric responses of all the modified electrodes investigated in this study for the oxidation of NO in 0.1 M phosphate buffer (pH 2.5) containing 1 mM of NO2− ions. The bare GCE- and GO-modified electrodes showed similar peak currents (Fig. 7a and b) at peak potentials of +0.82 V and +0.95 V, respectively. The rGO–Ag nanocomposite displayed a catalytic current response for the oxidation of 1 mM of NO irrespective of the amount of reducing agent with which it was prepared. Among all of the nanocomposites, the rGO–Ag (5.0 M) nanocomposite showed a higher catalytic current (94.6 μA) at a peak potential of +0.96 V because of the presence of a highly mono-dispersed Ag NPs with a smaller spherical size (Fig. 7e). The presence of a large population of smaller spherical Ag NPs with highly reduced GO increased with an increase in the ascorbic acid used as a reducing agent, which facilitated an efficient electron-transfer process during the electrocatalytic oxidation of NO and showed a synergetic catalytic current. However, the rGO–Ag (5.0 M) nanocomposite-modified electrode did not produce any enhanced voltammetric signal in the absence of NO (Fig. 7f). The rGO–Ag (0.5 M) nanocomposite produced less catalytic current (44.2 μA) at a peak potential of +0.91 V because of the presence of a lower population of larger Ag NPs with a more exposed GO surface (Fig. 5c). Compared to the rGO–Ag (0.5 M) nanocomposite, the rGO–Ag (1.0 M) nanocomposite showed a smaller difference in the peak current (35.7 μA), and the peak potential of NO was shifted toward a less positive potential (Fig. 7d). The graphical representation of the electrocatalytic oxidation of NO at the GC/rGO–Ag nanocomposite-modified electrode is shown in Scheme S1.† The stability of the GO–Ag (5.0 M) nanocomposite-modified electrode was checked by obtaining voltammograms using the same modified electrode for NO oxidation on different days. The voltammetric response showed only a 6.4% decrease in the oxidation current with a slight positive shift in the overpotential after seven days (Fig. S5†). This revealed the stability of the present modified electrode for NO oxidation. During stability measurements, the modified electrode was kept in a closed container at room temperature (25 °C).
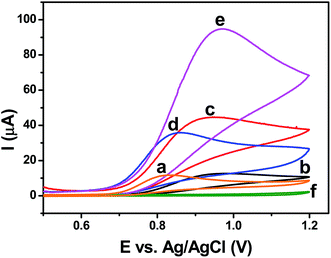 |
| | Fig. 7 Cyclic voltammograms recorded for 1 mM NO2− at bare GCE (a), GO (b), rGO–Ag (0.5 M) (c), 1.0 M (d), 5.0 M (e) nanocomposites modified electrodes in 0.1 M phosphate buffer (pH 2.5) with a scan rate of 50 mV s−1. (f) Cyclic voltammogram recorded at GO–Ag (5.0 M) nanocomposite modified electrode in the absence of NO2−. | |
The anodic peak current for the oxidation of NO increased with the NO concentration (Fig. S6†), and the plot of the peak current versus concentration shows a linear relation (Fig. S6 (inset)†). The effect of the scan rate on the oxidation of NO was studied at the rGO–Ag (5.0 M) nanocomposite-modified electrode (Fig. S7†). The oxidation peak current due to NO linearly increased with the scan rate. The peak current for NO oxidation showed a linear response in relation to the square root of the scan rate (ν1/2) (Fig. S7 (inset “A”)†) and this indicated that the electrocatalytic oxidation of NO at the rGO–Ag (5.0 M) nanocomposite-modified electrode was controlled by the diffusion process.43 The chemical irreversibility of the electrocatalytic NO oxidation process at the nanocomposite-modified electrode could be specified by a regular increase in the scan rate (ν) and increase in the oxidation peak potentials (Epa). The irreversible electrooxidation of NO was determined by the linear relation between log(ν) and the peak potential (Ep) (Fig. S7 (inset “B”)†). Chronoamperograms were recorded at the rGO–Ag (5.0 M) nanocomposite-modified electrode at different concentrations of NO2− ions (Fig. S8A†), and the plot of the peak current versus t−1/2 showed a linear relation (Fig. S8B†). The slopes of the obtained linear lines were plotted against the NO concentrations (Fig. S8B (inset)†), and based on the plot, the diffusion coefficient (D) of NO was calculated to be 6.470 × 10−5 cm2 s−1 using the Cottrell equation (eqn (2)).
| | |
I = nFD1/2AC0π−1/2t−1/2
| (2) |
where
n is the number of electrons transferred per NO molecule during oxidation,
F is the Faraday constant,
C0 is the concentration of NO,
A is the geometric area of the electrode, and
t is time.
3.4. Amperometric detection of NO
The sensing ability of the rGO–Ag (5.0 M) nanocomposite-modified electrode was investigated using the amperometric i–t curve technique with an applied potential of +0.96 V for the detection of 10 μM NO during the continuous stirring of 0.1 phosphate buffer (pH 2.5). Fig. 8A shows the amperometric i–t curve response for the detection of NO at the rGO–Ag (5.0 M) nanocomposite-modified electrode and the corresponding calibration plot obtained with every single addition of 10 μM NO in a homogeneously stirred solution of 0.1 M phosphate buffer. The current response increased linearly with the sequential injection of NO with a signal-to-noise (S/N) ratio of ∼3. The calibration plot of the current response (Id) difference and NO concentration showed a linear response in the concentration range of 10–220 μM, with a correlation coefficient R2 = 0.998 (y = 0.01065x + 1.0096 × 10−8), and the sensitivity of the rGO–Ag (5.0 M) nanocomposite electrode was found to be 0.01065 μA μM−1 for the detection of NO (Fig. 8A (inset)). The LOD for the detection of NO was calculated to be 2.84 μM using the formula LOD = 3.3(SD/slope), where SD is the standard deviation of the y-intercepts.44
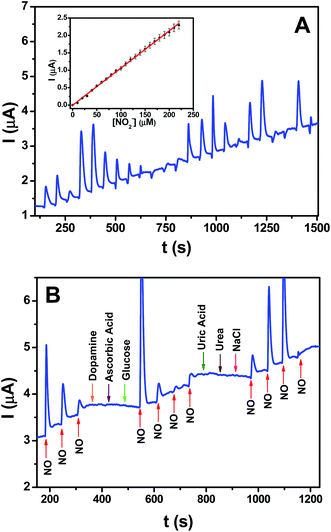 |
| | Fig. 8 (A) Amperometric i–t curve of GC/rGO–Ag (5.0 M) nanocomposite modified electrode for each addition of 10 μM NO2− in 0.1 M phosphate buffer (pH 2.5) at a regular time interval of 60 s (applied potential was +0.96 V). Inset: plot of current versus concentration of NO2−. (B) Amperometric i–t curve of GC/rGO–Ag nanocomposite modified electrode for the addition of 10 μM NO2− and each 100 μM addition of other interferents in 0.1 M phosphate buffer (pH 2.5) at a regular time interval of 60 s (applied potential was +0.96 V). | |
3.5. Interference analysis
The selectivity of the rGO–Ag (5.0 M) nanocomposite for NO detection was examined by injecting various possible common interferents such as ascorbic acid, dopamine, glucose, urea, uric acid, and sodium chloride (NaCl) in the phosphate buffer containing NO under continuous stirring, and then observing the change in the current response (Fig. 8B). From the i–t curve, it was found that the current signal only appeared when injecting the NO into the phosphate buffer, with no current response appearing even when injecting a 10-fold higher concentration of interferents than the concentration of NO. Furthermore, after the highest concentration of interfering ions was added, the current signals corresponding to the addition of NO were reproduced with nearly the same magnitudes. These results showed the selectivity of NO at the GC–rGO–Ag (5.0 M) nanocomposite-modified electrode. Table 1 shows a comparison of the analytical performances of the present nanocomposite and some of the reported materials for NO detection. In addition to the appreciable LOD and selectivity, the novelty of the present work lies in the easy deposition of Ag NPs on rGO sheets using ascorbic acid as a reducing agent, and the systematic investigation of the electrochemical properties for NO oxidation and in situ detection. The in situ reduction of both Ag+ ions and the oxygen functionalities of GO by ascorbic acid was achieved. The cost of the electrochemical sensor for NO detection could be reduced with the use of Ag metal, and this is a good alternate for other electrochemical sensors prepared with noble metals like Au, Pt, and Pd and expensive polymeric materials.
Table 1 Comparison of analytical performance of some of the reported sensor electrodes with the present nanocomposite for NO detectiona
| Electrode material |
Analytical method |
Linear range (M) |
Limit of detection (M) |
Interference studied |
Ref. |
| DPV = differential pulse voltammetry; SWV = square wave voltammetry; ERG = electrochemically reduced graphene; ERGO = electrochemically reduced graphene oxide; AuNPs = gold nanoparticles; CeO2 = ceria; G–Nf = graphene–nafion; CO3O4@Pt = cobalt oxide nanocube@platinum; Au–TPDT NRs = gold nanorods embedded in an amine functionalized silicate sol–gel matrix; PtNP = platinum nanoparticles; AB = acetylene black; Hb = haemoglobin; CPB = cetylpyridinium bromide; PAM = polyacrylamide; G–Au = graphene–gold; PNMP-b-PGMA = poly[N-(2-methacryloyloxyethyl)pyrrolidone]-block-poly[glycidyl methacrylate]; PAM = polyacrylamide; SDS = sodium dodecyl sulfate; Cyt c= cytochrome c; AA = ascorbic acid; UA = uric acid; NaCl = sodium chloride. |
| GCE/rGO–Co3O4@Pt |
Amperometry |
10 × 10−6 to 650 × 10−6 |
1.73 × 10−6 |
Dopamine, AA, UA, urea, glucose, NaCl |
41 |
| GCE/ERG |
Amperometry |
7.2 × 10−7 to 7.8 × 10−5 |
2.0 × 10−7 |
AA |
45 |
| GCE/AuNPs–ERGO |
Amperometry |
Up to 3.38 × 10−6 |
1.33 × 10−7 |
Oxalate, glucose, UA, NaCl, AA |
46 |
| GCE/rGO–CeO2 |
Amperometry |
18.0 × 10−9 to 5.6 × 10−3 |
9.6 × 10−9 |
— |
47 |
| GCE/G–Nf |
SWV |
0.05 × 10−3 to 0.45 × 10−3 |
11.61 × 10−6 |
— |
48 |
| GCE/RGO–Au–TPDT NRs |
Amperometry |
10 × 10−9 to 140 × 10−9 |
6.5 × 10−9 |
Glucose, urea, oxalate, NaCl |
49 |
| GCE/PtNP/AB |
Amperometry |
0.18 × 10−6 to 120 × 10−6 |
0.05 × 10−6 |
Glucose, caffeine, D-L-valine, L-glutamic acid, L-aspartic acid, glycine, cholesterol, L-serine and barbitone, L-arginine, L-tyrosine, sucrose, L-cystine, UA, AA, NO2− |
50 |
| GCE/Hb–CPB/PAM |
CV |
9.8 × 10−6 to 100 × 10−6 |
9.3 × 10−6 |
— |
51 |
| GCE/G–Au |
Amperometry |
1 × 10−6 to 1100 × 10−6 |
0.25 × 10−6 |
H2O2, dopamine, glucose, AA, UA |
52 |
| GCE/PNMP-b-PGMA/Hb |
DPV |
0.45 × 10−6 to 10 × 10−6 |
0.32 × 10−6 |
— |
53 |
| GCE/PAM/SDS/Cyt c |
Amperometry |
0.80 × 10−6 to 95 × 10−6 |
0.1 × 10−6 |
K+, Na+, NH4+, Mg2+, Al3+, Ca2+, Cu2+, SO42−, CO32−, NO3−, Cl− |
54 |
| PGE/Hb–silver NPs |
CV |
1.0 × 10−6 to 5.0 × 10−5 |
3.0 × 10−7 |
Ascorbate, catechol, dopamine, epinephrine, nitrite, UA |
55 |
| GCE/rGO–Ag |
Amperometry |
10 × 10−6 to 220 × 10−6 |
2.84 × 10−6 |
Dopamine, AA, UA, urea, glucose, NaCl |
This work |
3.6. Real sample analysis
The detection of NO was performed using tap and lake water samples to validate the applicability of the rGO–Ag nanocomposite for practical samples. The obtained good recoveries of three different spiked concentrations of NO2− revealed that the sensor could be used for the in situ detection of NO in environmental water samples. Each experiment was repeated three times and the mean % recovery was determined (Table 2).
Table 2 Measurement results of NO in real water samples
| Real samples |
Concentration spiked (μM) |
Concentration found (μM) |
Recovery (%) |
RSD (%) |
| Tap water |
10 |
9.5 |
95.0 |
3.0 |
| 50 |
53.1 |
106.2 |
1.9 |
| 100 |
104.6 |
104.6 |
1.5 |
| Lake water |
10 |
9.6 |
96.0 |
1.3 |
| 50 |
53.8 |
107.6 |
2.1 |
| 100 |
105.5 |
105.5 |
1.6 |
4. Conclusions
In this work, we verified the one-pot synthesis of Ag NPs on rGO sheets using the chemical reduction of AgNO3 in the presence of a GO suspension and ascorbic acid as a reducing. The concentration of ascorbic acid influenced the size of the Ag NPs the reduction GO. The formation of smaller spherical (average size of 2 nm) and highly crystalline nanoparticles was observed in the rGO–Ag nanocomposite with 5.0 M ascorbic acid. An electrochemical system was constructed using the rGO–Ag nanocomposite to study its electrochemical properties for NO oxidation. The rGO–Ag (5.0 M) nanocomposite-modified GC electrode showed a better catalytic response in cyclic voltammetry for NO oxidation, and the reaction followed a diffusion-controlled process at the modified electrode surface. The modified electrode was used for the in situ detection of NO in 0.1 M phosphate buffer (pH 2.5), and the LOD was found to be 2.84 μM using an amperometry technique. The rGO–Ag (5.0 M) nanocomposite was verified to have good stability and better selectivity among physiological interferents such as uric acid, ascorbic acid, urea, dopamine, glucose, and NaCl. The applicability of the present sensor was verified in environmental water samples and good recoveries were found.
Acknowledgements
The authors gratefully acknowledge a Postgraduate Research Grant (PG134-2015B) by the University of Malaya and the staff of the photoemission spectroscopy (PES) synchrotron beamlines 3.2a at the Synchrotron Light Research Institute, Thailand, for their technical assistance and beamline support. Nurul Izrini Ikhsan would like to thank the Ministry of Higher Education (MOHE) and Mara University of Technology (UiTM), Shah Alam for the scholarship they sponsored.
References
- Y. M. Liu, C. Punckt, M. Pope, A. Gelperin and I. Aksay, ACS Appl. Mater. Interfaces, 2013, 5, 12624 CAS.
- F. L Ricciardolo, P. J. Sterk, B. Gaston and G. Folkerts, Physiol. Rev., 2004, 84, 731 CrossRef PubMed.
- C. Napoli and L. J. Ignarro, Nitric Oxide, 2001, 5, 88 CrossRef CAS PubMed.
- O. Traub and R. Van Bibber, West. J. Med., 1995, 162, 439 CAS.
- P. Thejass and G. Kuttan, Nitric Oxide, 2007, 16, 247 CrossRef CAS PubMed.
- R. Kavya, R. Saluja, S. Singh and M. Dikshit, Nitric Oxide, 2006, 15, 280 CrossRef CAS PubMed.
- Y. M. Liu, C. Punckt, M. A. Pope, A. Gelperin and I. A. Aksay, ACS Appl. Mater. Interfaces, 2013, 5, 12624 CAS.
- L. Zhang, Y. Ni, X. Wang and G. Zhao, Talanta, 2010, 82, 196 CrossRef CAS PubMed.
- L. A. Ridnour, J. E. Sim, M. A. Hayward, D. A. Wink, S. M. Martin, G. R. Buettner and D. R. Spitz, Anal. Biochem., 2010, 281, 223 CrossRef PubMed.
- E. M. Hetrick and M. H. Schoenfisch, Annu. Rev. Anal. Chem., 2009, 2, 409 CrossRef CAS PubMed.
- X. Ye, S. S. Rubakhin and J. V. Sweedler, Analyst, 2008, 133, 423 RSC.
- P. Wu, J. Wang, C. He, X. Zhang, Y. Wang, T. Liu and C. Duan, Adv. Funct. Mater., 2012, 22, 1698 CrossRef CAS.
- A. S. Adekunle, S. Lebogang, P. L. Gwala, T. P. Tsele, L. O. Olasunkanmi, F. O. Esther and E. E. Ebenso, RSC Adv., 2015, 5, 27759 RSC.
- T. Endo, K. Kerman, N. Nagatani, H. M. Hiepa, D. K. Kim, Y. Yonezawa and E. Tamiya, Anal. Chem., 2006, 78, 6465 CrossRef CAS PubMed.
- Y. M. Liu, C. Punckt, M. A. Pope, A. Gelperin and I. A. Aksay, ACS Appl. Mater. Interfaces, 2013, 5, 12624 CAS.
- A. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425 RSC.
- J. Greeley, I. E. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- G. Zhang and M. Liu, Sens. Actuators, B, 2000, 69, 144 CrossRef CAS.
- Q. Wang, H. Yu, L. Zhong, J. Liu, J. Sun and J. Shen, Chem. Mater., 2006, 18, 1988 CrossRef CAS.
- T. Dadosh, Mater. Lett., 2009, 63, 2236 CrossRef CAS.
- X. Zan and Z. Su, Langmuir, 2009, 25, 12355 CrossRef CAS PubMed.
- Q. Qu, S. Yang and X. Feng, Adv. Mater., 2011, 23, 5574 CrossRef CAS PubMed.
- P. V. Kamat and J. Phys, Chem. Lett., 2009, 1, 520 Search PubMed.
- Y. Q. Guo, X. P. Sun, Y. Liu, W. Wang, H. X. Qiu and J. P. Gao, Carbon, 2012, 50, 2513 CrossRef CAS.
- Y. W. Zhang, S. Liu, L. Wang, X. Y. Qin, J. Q. Tian, W. B. Lu, G. H. Chang and X. P. Sun, RSC Adv., 2012, 2, 538 RSC.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279 CrossRef CAS PubMed.
- S. Villar-Rodil, J. I. Paredes, A. Martínez-Alonso and J. M. Tascón, J. Mater. Chem., 2009, 19, 3591 RSC.
- N. Huang, H. Lim, C. Chia, M. Yarmo and M. Muhamad, Int. J. Nanomed., 2011, 6, 3443 CrossRef CAS PubMed.
- S. Xu, L. Yong and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 654 CAS.
- M. Liu, M. Leng, C. Yu, X. Wang and C. Wang, Nano Res., 2010, 3, 843 CrossRef CAS.
- J. I. Hussain, A. Talib, S. Kumar, S. A. AL-Thabaiti, A. A. Hashmi and Z. Khan, Colloids Surf., A, 2011, 381, 23 CrossRef CAS.
- W. Lu, G. Chang, Y. Luo, F. Liao and X. Sun, J. Mater. Sci., 2011, 46, 5260 CrossRef CAS.
- J. Jana, S. S. Gauri, M. Ganguly, S. Dey and T. Pal, Dalton Trans., 2015, 44, 20692 RSC.
- S. Radhakrishnan, K. Krishnamoorthy, C. Sekar, J. Wilson and S. J. Kim, Appl. Catal., B, 2014, 22, 148 Search PubMed.
- D. A. Dinh, K. S. Hui, K. N. Hui, Y. R. Cho, W. Zhou, X. Hong and H. H. Chun, Appl. Surf. Sci., 2014, 298, 62 CrossRef CAS.
- X. H. Meng, X. Shao, H. Y. Li, F. Z. Liu, X. P. Pu, W. Z. Li and C. H. Su, Mater. Res. Bull., 2013, 48, 1453 CrossRef CAS.
- C. Xu, X. Wang and J. W. Zhu, J. Phys. Chem. C, 2008, 112, 19841 CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H. M. Cheng, Carbon, 2010, 48, 4466 CrossRef CAS.
- Y. Zhang, X. Yuan, Y. Wang and Y. Chen, J. Mater. Chem., 2012, 22, 7245 RSC.
- N. I. Ikhsan, P. Rameshkumar, A. Pandikumar, M. M. Shahid, N. M. Huang, S. V. Kumar and H. N. Lim, Talanta, 2015, 144, 908 CrossRef CAS PubMed.
- M. M. Shahid, P. Rameshkumar, A. Pandikumar, H. N. Lim, Y. H. Ng and N. M. Huang, J. Mater. Chem. A, 2015, 3, 14458 CAS.
- S. Thangavel and R. Ramaraj, J. Phys. Chem. C, 2008, 112, 19825 CAS.
- M. A. Kamyabi and F. Aghajanloo, J. Electroanal. Chem., 2008, 614, 157 CrossRef CAS.
- S. S. Razola, B. L. Ruiz, N. M. Diez, H. B. Mark and J. M. Kauffmann, Biosens. Bioelectron., 2002, 17, 921 CrossRef CAS PubMed.
- Y.-L. Wang and G.-C. Zhao, Int. J. Electrochem., 2011, 2011, 1 Search PubMed.
- S. L. Ting, C. X. Guo, K. C. Leong, D.-H. Kim, C. M. Li and P. Chen, Electrochim. Acta, 2013, 111, 441 CrossRef CAS.
- F. X. Hu, J. Le Xie, S. J. Bao, L. Yu and C. M. Li, Biosens. Bioelectron., 2015, 70, 310 CrossRef CAS PubMed.
- N. Yusoff, A. Pandikumar, A. R. Marlinda, N. M. Huang and H. N. Lim, Anal. Methods, 2015, 7, 3537 RSC.
- S. Jayabal, P. Viswanathan and R. Ramaraj, RSC Adv., 2014, 4, 33541 RSC.
- D. Zheng, X. Liu and D. Zhou, Microchim. Acta, 2012, 55 Search PubMed.
- X. He and L. Zhu, Electrochem. Commun., 2006, 8, 615 CrossRef CAS.
- R. Geetha, K. Muthoosamy, M. Zhou and M. Ashokkumar, Biosens. Bioelectron., 2017, 87, 622 CrossRef PubMed.
- S. Jia, J. Fei, J. Deng, Y. Cai and J. Li, Sens. Actuators, B, 2009, 138, 244 CrossRef CAS.
- X. Chen, H. Long, W. Wu and Z. Yang, Thin Solid Films, 2009, 517, 2787 CrossRef CAS.
- X. Gan, T. Liu, X. Zhu and G. Li, Anal. Sci., 2004, 20, 1271 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21716h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.