
Quantum chemistry calculations of technetium and rhenium compounds with application in radiopharmacy: review
Abstract
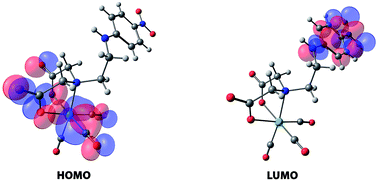
In the last 15 years, computational quantum chemistry has become an invaluable tool that supports the interpretation of experimental measurements of a broad range of molecular properties of Tc and Re compounds. Among the contemporary computational techniques, density functional theory is the most extended one. This review describes recent computational investigations that illustrate best the promise of quantum chemical calculation in a number of areas of Tc and Re chemistry, such as geometry, stability of complexes, molecular spectroscopic properties and electronic structures and bonding characters. General trends and the prospects for future applications are also discussed.
 Please wait while we load your content...
Please wait while we load your content...

