
Dieckol, an edible seaweed polyphenol, retards rotenone-induced neurotoxicity and α-synuclein aggregation in human dopaminergic neuronal cells†
 *abe
*abe
Abstract
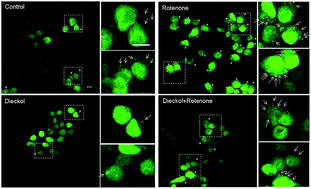
Dopaminergic neurons are particularly vulnerable to oxidative stress, which may initiate a cascade of intracellular toxic events that lead to protein aggregation and subsequent cell death, causing Parkinson's disease. Here, we investigate the neuroprotective effect of dieckol, which is a polyphenol isolated from an edible seaweed, Ecklonia cava, on rotenone-induced oxidative stress in SH-SY5Y cells, a human dopaminergic neuronal cell line. Dieckol was found to reduce intracellular reactive oxygen species (ROS) and cytochrome C release induced by treatment with rotenone. Consequently, dieckol reduced rotenone-induced cell death, and retarded rotenone-induced α-synuclein aggregation in α-synuclein-overexpressing SH-SY5Y cells. These results clearly indicate that dieckol possesses prominent antioxidant activity in dopaminergic neuronal cells preventing α-synuclein aggregation. Therefore, it could be a potential therapeutic agent for the prevention of neurodegenerative diseases such as Parkinson's disease.
 Please wait while we load your content...
Please wait while we load your content...

