
Dual release of a hydrophilic and a hydrophobic osteogenic factor from a single liposome
Nelson Monteiro ab,
Albino Martinsab,
Ricardo A. Piresab,
Susana Fariac,
Nuno A. Fonsecad,
João N. Moreirad,
Rui L. Reisab and
Nuno M. Neves*ab
ab,
Albino Martinsab,
Ricardo A. Piresab,
Susana Fariac,
Nuno A. Fonsecad,
João N. Moreirad,
Rui L. Reisab and
Nuno M. Neves*ab
a3B's Research Group – Biomaterials, Biodegradables and Biomimetics, Department of Polymer Engineering, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark, Zona Industrial da Gandra S. Cláudio do Barco, 4806-909 Caldas das Taipas, Guimarães, Portugal. E-mail: nelson.monteiro@gmail.com; amartins@dep.uminho.pt; rpires@dep.uminho.pt; rgreis@dep.uminho.pt; nuno@dep.uminho.pt
bICVS/3B's, PT Government Associate Laboratory, Braga, Guimarães, Portugal
cResearch Center Officinal Mathematical, Department of Mathematics for Science and Technology, University of Minho, Campus de Azurém, 4800-058 Guimarães, Portugal. E-mail: sfaria@math.uminho.pt
dCenter for Neurosciences and Cell Biology (CNC), Faculty of Pharmacy of the University of Coimbra, 3000 Coimbra, Portugal. E-mail: nuno.a.c.fonseca@gmail.com; jmoreira@ff.uc.pt
First published on 24th November 2016
Abstract
Delivery systems may be designed to protect and control the release kinetics of growth/differentiation factors in a spatiotemporal manner. Liposomes are examples of biological-based bioactive agent delivery systems. In this work, ascorbic acid (AscA) was encapsulated in the inner compartment of the liposome and dexamethasone (Dex) was encapsulated within the lipid bilayer in order to develop a dual release system of these bioactive agents involved in the osteogenic differentiation of mesenchymal stem cells (MSCs). The particle size (∼150 nm) of the prepared liposomes showed a monodisperse distribution. The bioactive agent release study showed that Dex was released more rapidly from the liposomes than AscA. The Dex release profile showed an initial burst release within 12 h; afterwards, a slower and sustained release was observed until 21 days. The release of AscA from the liposomes was not detected until 6 h; afterwards, a linear release was observed from 24 h until 21 days. The effect of Dex–AscA-loaded liposomes on the viability, proliferation and osteogenic differentiation of human bone marrow-derived MSCs (hBMSCs) were assessed. The cell culture results showed that the Dex–AscA-loaded liposomes (in a single dose or in repeated doses) do not have any cytotoxic effect. Dex–AscA-loaded liposomes given once did not promote induction of hBMSCs differentiation into the osteogenic lineage. However, Dex–AscA-loaded liposomes given repeatedly promoted the hBMSCs differentiation into the osteogenic lineage, both in basal medium and complete osteogenic medium. These results were genotypically demonstrated by the expression of osteoblastic markers. In conclusion, Dex–AscA-loaded liposomes represent a biological nanoparticle strategy with potential safety and efficacy for bone tissue engineering approaches.
Introduction
The natural regeneration or healing of tissues is a highly complex cascade of events, involving several biomolecules that act synergistically and are mediated by cross-talk mechanisms.1,2 Thus, the delivery of individual bioactive factors may not be able to promote the desired bioactivity.3–5 Therefore, the need for regulation of tissue regeneration by bioactive agents has been eliciting the use and fabrication of nano-devices, such as nanoparticles and nanofibers, for the delivery of multiple bioactive agents.6,7 Bioactive agent delivery systems are designed to stimulate, respond to and interact with target cells and tissues at the scale of those physiological events.8,9 Moreover, encapsulation technology has proven to be an excellent tool for Tissue Engineering and Regenerative Medicine approaches, providing opportunities for the researchers to develop novel formulations with improved functionalities.10 Examples of delivery systems include liposomes, nanospheres, nanocapsules, micelles and dendrimers.11–14 The main advantages of using nanoparticles are the capacity to diffuse through cytoplasmatic cell membranes and the ability to be guided to a specific cell type through the use of specific ligands. Furthermore, the use of nanoparticles may provide several additional advantages such as the possibility to deliver multiple bioactive agents simultaneously and to create spatial and temporal patterns of bioactive agent release patterns.15,16Liposomes are lipid-based carrier systems.17 They have been widely used to deliver drugs into pathological sites such as tumors and inflammatory regions, and as vehicles for gene delivery.18–20 Moreover, they are very versatile since they enable encapsulation of both hydrophilic and lipophilic compounds.21–24 Lipophilic drugs are encapsulated within the lipid bilayer of the liposome (the non-polar, hydrophobic compartment) while hydrophilic drugs are encapsulated in the inner core of the liposome (the polar compartment). Liposomes also present significant advantages over other nanoparticle-based drug release systems such as a high load carrying capacity,25 low cytotoxicity26 and a versatile structure in terms of possible formulations and functionalization.27 Many attempts on the use of liposomes for dual release of bioactive agents have been proposed.18,28–30 For instance, double liposomes consist of several small liposomes encapsulated into a large liposome, being the outer liposomes able to protect the inner liposomes against enzyme degradation.29 Therefore, one drug can be encapsulated into the inner liposomes, and another drug can be encapsulated into the outer liposomes.18 The main disadvantage of this system relies on the fact that the large liposomes are rapidly eliminated by macrophages and cannot diffuse through cytoplasmatic cell membranes.
Human mesenchymal stem cells (hMSCs) are adult stem cells that are found in specific tissues of the body such as bone marrow, adipose tissue, teeth, dermis and umbilical cord blood.31–33 They have the potential of self-renewal, expansion and differentiation into various mesodermal tissues, i.e. bone, cartilage, fat, muscle and other connective tissues.34 Therefore, they are excellent cell candidates for autologous tissue regeneration therapies.35–38 However, after isolation and ex vivo expansion, the challenges rely in controlling the MSCs differentiation and deliver them at the target site.39 Many strategies to differentiate MSCs into the desired lineages have been proposed.40,41 The classical method for the in vitro osteogenic differentiation of MSCs involves supplementation of culture medium with bioactive agents including dexamethasone (Dex), beta-glycerophosphate (β-GP) and ascorbic acid (AscA).36 Moreover, other combinations of bioactive agents could be used such as vitamin D3 (vitD3), transforming growth factor-beta (TGFb) and bone morphogenetic proteins (BMPs).32
In the present work, we develop a dual bioactive agent-loaded liposome, using the space within the lipid bilayer and the inner core of the liposome, as a new method to induce the differentiation of MSCs. Specifically, Dex was encapsulated within the lipid bilayer, and AscA encapsulated in the inner compartment of the liposome. Dex is a lipophilic corticosteroid that has shown capabilities to induce the differentiation of MSCs.32,36,42–44 However, at high concentrations exert inhibitory effects over MSCs proliferation and matrix collagen formation.32,45 AscA or vitamin C is physiologically and biochemically an essential water-soluble bioactive compound required for normal regulatory functions.46 AscA is used for the synthesis of collagen and other organic components of the extracellular matrix of some tissues such as bones, skin and other connective tissues.32,47 The encapsulation of Dex and AscA in the liposomes may provide their long-term stability and bioactivity and may enhance the osteogenic differentiation of human bone marrow-derived MSCs (hBMSCs) in vitro. We studied the effect of this dual release system in basal medium (absence of typical osteogenic cocktails) and in the osteogenic differentiation medium (medium with β-GP), in a single dose and in repeated doses.
Experimental section
Materials
Chloroform, ascorbic acid (AscA) dexamethasone (Dex), ammonium molybdate, fiske–subbarow reducer, sepharose CL4B and HEPES (HEPES buffer solution, HBS) were purchased from Sigma-Aldrich. The lipids L-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Egg-transphosphatidylated, chicken) (PE-Rho), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE–PEG), cholesterol (ovine wool, >98%) (Chol) and L-α-phosphatidylcholine, hydrogenated (Soy) (HSPC) were purchased from Avanti Polar Lipids. Dialysis tubing cellulose membrane (100–500 molecular weight cut off, 10 mm flat width) was purchased from Spectrum Labs. Ascorbic acid (AscA) quantification kit (K-ASCO) was purchased from Megazyme. All the materials were used as received.Production and characterization of Dex–AscA-loaded liposomes
The liposome formulations used in this study are presented in Table 1. Formulations A, B and C were used to study the encapsulation efficiency of AscA and Dex in the liposomes, varying the AscA concentration (i.e. 10, 25 and 50 mg) and keeping the Dex molar ratio constant as optimized in a previous study.48 Formulation D was used to perform the AscA and Dex release study from the liposomes. Formulation E and F was used for the biological assays.| AscA (mg) | Dex | HSPC | DSPE–PEG | PE–Rho | |
|---|---|---|---|---|---|
| A | 10 | 0.25 | 2 | — | — |
| B | 25 | 0.25 | 2 | — | — |
| C | 50 | 0.25 | 2 | — | — |
| D | 50 | 0.25 | 2 | 0.1 | — |
| E | 0.25 | 2 | 0.1 | 0.02 | |
| F | 50 | 0.25 | 2 | 0.1 | 0.02 |
The production of Dex-loaded liposomes was performed as reported elsewhere in detail.48 Briefly, lipids and Dex were mixed in a round-bottom flask, using proper amounts of each lipid (15 mM total lipid in chloroform), in the proportions described for each type of formulation. The solvent was slowly evaporated using a gentle stream of nitrogen. The obtained dry film was dispersed, using vortex, in a solution of AscA in HBS (at pH 5), keeping the temperature of the hydrating medium above the gel–liquid crystal transition temperature (Tc = 52 °C). The multilamellar liposomal suspension was extruded at T > Tc using a porous polycarbonate membrane (pore size of 100 nm) held between two tight syringes. The syringes were used to force the solution back and forth (21 times), resulting in unilamellar liposome vesicles. Non-encapsulated Dex and AscA were removed from the solution by column chromatography (Sepharose CL4B, Sigma-Aldrich) using an isocratic elution with HBS at pH 7.4. Particle size distribution and ζ-potential were determined by dynamic light scattering (Zetasizer Nanoseries ZS, Malvern Instruments).
Dex and AscA loading efficiency into liposomes
The Dex and AscA loading efficiencies were determined using the formulations A, B and C, by quantifying their Dex, AscA and lipid content. Dex concentration was determined by high performance liquid chromatography (HPLC, KNAUER) using an Atlantis T3 5 μm C-18 column, a flow rate of 1 mL min−1, 0.2% phosphoric acid in a water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile (55:45) mixture, as the mobile phase, and UV detection at 247 nm. Dex was quantified using a calibration curve obtained with standard solutions. AscA concentration was determined by the K-ASCO kit as described by the manufacturer using the microplate formats. Basically, dehydroascorbic acid was converted to ascorbic acid by the addition of DTT to the samples at a final concentration of 1 mM, followed by incubation at room temperature during 10 minutes. The difference in the AscA content of the DTT-treated sample compared to the untreated sample is proportional to the dehydroascorbic acid content. The absorbance was measured using a microplate reader (Synergie HT) at 578 nm. The total lipid concentration was assessed by the Bartlett colorimetric assay as described elsewhere in detail.49 The principle of the Bartlett assay is based on the colorimetric determination of inorganic phosphate resulting from the transformation of the phospholipids of liposomes using perchloric acid. The inorganic phosphate is converted to phospho-molybdic acid by the addition of ammonium molybdate, which is reduced to a blue colored complex by 4-amino-2-naphthyl-4-sulfonic acid during heating. This compound can be determined colorimetrically at 830 nm.
:acetonitrile (55:45) mixture, as the mobile phase, and UV detection at 247 nm. Dex was quantified using a calibration curve obtained with standard solutions. AscA concentration was determined by the K-ASCO kit as described by the manufacturer using the microplate formats. Basically, dehydroascorbic acid was converted to ascorbic acid by the addition of DTT to the samples at a final concentration of 1 mM, followed by incubation at room temperature during 10 minutes. The difference in the AscA content of the DTT-treated sample compared to the untreated sample is proportional to the dehydroascorbic acid content. The absorbance was measured using a microplate reader (Synergie HT) at 578 nm. The total lipid concentration was assessed by the Bartlett colorimetric assay as described elsewhere in detail.49 The principle of the Bartlett assay is based on the colorimetric determination of inorganic phosphate resulting from the transformation of the phospholipids of liposomes using perchloric acid. The inorganic phosphate is converted to phospho-molybdic acid by the addition of ammonium molybdate, which is reduced to a blue colored complex by 4-amino-2-naphthyl-4-sulfonic acid during heating. This compound can be determined colorimetrically at 830 nm.
The ability of the liposomal vesicles to incorporate Dex and AscA was evaluated by calculating the system payload (PL) from eqn (1). The encapsulation efficiency (EE) was calculated as the final PL (PLf) per initial PL (PLi) of Dex or AscA (mM) and HSPC (mM) from eqn (2).
 | (1) |
 | (2) |
Dex and AscA release kinetics from the liposomes
The release of Dex and AscA from the loaded liposomes was studied using a dialysis method. Dialysis cellulose tubes (100–500 molecular weight cut off, Spectrum Labs) were rinsed with distilled water for more than one week prior to their use. Tube ends were closed with Teflon tape and Nylon thread and tested for leakage. 1 mL of liposomal solution (formulation D) was added to the dialysis tubes and fully immersed into 10 mL of PBS as release medium. Control tubes were assembled using the same procedure with Dex–AscA free liposomes. The solutions were maintained at 37 °C and 60 rpm during 21 days. At defined time points, an aliquot of 1 mL was collected from the solution and the Dex and AscA concentration assessed as described above. The same volume of fresh PBS was replaced to keep the total volume of release medium at 10 mL. The experiments were performed in triplicate.The study the of drug release mechanism used the equations eqn (3) and (4) to model the Dex and the AscA release kinetics. The data was fitted with the equations using a non-linear regression in the case of Dex release (eqn (3)) and a linear regression in the case of the AscA release (eqn (4)).
| C = a0 + ktn | (3) |
| C = a0 + kt | (4) |
The power law equation (eqn (3)) was selected because it is the most widely used equation in drug release studies,50 where C is the cumulative release of the drug at the time t, a0 is a constant representing the percentage of burst release, k is the kinetic constant and n is the release exponent, indicating the mechanism of drug release.
Biological assays
Expansion, seeding and osteogenic differentiation of human bone marrow mesenchymal stem cells
hBMSCs were isolated from bone marrow aspirates collected under informed consent from patients undergoing knee arthroplasty at the Hospital de Braga, Portugal, in accordance to the protocol established between the 3B's Research Group and the Hospital de Braga and approved by the ethics committee of the same Hospital. Samples were collected from a 58 year old female donor, isolated, expanded and cryopreserved until further use. hBMSCs were isolated and characterized according to the method established by Delorme et al.51 Briefly, plastic adherent fractions of marrow cells characterized by a spindle-shape morphology and colony-forming unit (CFU) capacity; positive expression of the surface antigens CD 29, 73, 90 and 105, and negative for hematopoietic markers such as CD 34 and 45 (all the antibodies were purchased from BD Pharmingen (USA), and the hBMSCs were analyzed on a FACS Calibur, BD Biosciences (USA)); and by their differentiation potential into the osteogenic, chondrogenic and adipogenic lineages. hBMSCs were expanded in basal medium consisting of MEM alpha medium (α-MEM; Gibco, GB) supplemented with 10% heat-inactivated fetal bovine serum (BiochromAG, Germany) and 1% antibiotic/antimyotic solution (final concentration of penicillin 100 units per mL and streptomycin 100 mg mL−1; Gibco, GB). The cells were cultured at 37 °C in a humidified atmosphere of 5% CO2, and the medium was exchanged every 2–3 days. The liposomal suspension (formulation E) was sterilized by 0.22 μm filters. Confluent hBMSCs at passage 4 were harvested for seeding onto 24 well plates at a density of 1 × 105 cells per well. After 24 h of incubation in basal medium, hBMSCs were cultured in basal (absence of typical osteogenic cocktail) and in standard osteogenic differentiation medium (basal medium supplemented with 10−7 M Dex, 50 μg mL−1 AscA and 10 mM β-GP). At the same time, hBMSCs were cultured in the presence of Dex-loaded liposomes (single dose and repeated in basal and osteogenic medium with AscA and β-GP, i.e. culture conditions E(once)_Basal, E(repeat)_Basal, E(once)_Osteo, E(repeat)_Osteo) and Dex–AscA-loaded liposomes (single dose and repeated in basal and osteogenic medium with β-GP, i.e. culture conditions F(once)_Basal, F(repeat)_Basal, F(once)_Osteo and F(repeat)_Osteo). The cells were retrieved at predefined culturing times: 7, 14 and 21 days. The experiments were performed three times independently, with sample triplicates.Cell viability and proliferation assessment
Cell viability for each culturing condition and time point was determined using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, USA), according to the manufacturer instructions. This assay is based on the bioreduction of a tetrazolium compound, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfofenyl)-2H-tetrazolium [MTS], into a water-soluble brown formazan product. The absorbance was measured at 490 nm in a microplate reader (SynergieHT, Bio-Tek, USA), being related with the quantity of formazan product and directly proportional to the number of living cells in culture.Cell proliferation was quantified by the total amount of double-stranded DNA, along the culturing time. Quantification was performed using the Quant-iT™ Pico-Green dsDNA Assay Kit (Invitrogen™, Molecular Probes™; Oregon, USA), according to the instructions of the manufacturer. Briefly, cells were lysed by osmotic and thermal shock and the supernatant used for the DNA quantification assay. The fluorescence of the dye was measured at an excitation wavelength of 485/20 nm and an emission wavelength of 528/20 nm, in a microplate reader (Synergie HT, Bio-Tek; USA). Quadruplicates were made for each sample and per culturing time. The DNA concentration for each sample was calculated using a standard curve (DNA concentration ranging from 0.0 to 1.5 μg mL−1) relating the DNA quantity to the fluorescence intensity.
All data concerning cell viability, total protein and ALP activity were independently measured and normalized against the cell number for each sample. For that, a standard calibration curve was performed using known hBMSCs cell numbers at passage 4, ranging from 0 to 5 × 105 cells, with an n = 12. The dsDNA concentration of these samples was determined according to the Quant-iT™ PicoGreen® dsDNA Reagent, previously described. The following equation was obtained: y = 0.0054x + 86.68 with a R2 = 0.998, where the y is the measured fluorescence value and the x is the cell number, and used to estimate the cell numbers for each measurement.
Alkaline phosphatase and total protein quantification
The concentration of alkaline phosphatase (ALP) was determined for all time culture periods, using the lysates used for DNA quantification. Briefly, the ALP quantity was assessed using the p-nitrophenol assay, in which 4-nitrophenyl phosphate disodium salt hexahydrate (Sigma, USA) is hydrolysed by the intracellular ALP at the temperature of 37 °C in an alkaline buffer solution (1.5 M and pH 10.5, Sigma) to form yellow free p-nitrophenol. The reaction was stopped by the addition of 0.3 M NaOH (Panreac Quimica, Spain) and the absorbance read at 405 nm in a microplate reader (Bio-Tek, Synergie HT, USA). Standards were prepared with 10 mM p-nitrophenol (pNP; Sigma, USA) solution, to obtain a standard curve ranging from 0 to 250 μM. Quadruplicates of each sample and standard were made, and the ALP concentrations were obtained from the standard curve.For the quantification of total protein synthesized by the hBMSCs in culture, the Micro BCA™ Protein Assay kit (Thermo Scientific, Pierce; Rockford, USA) was used according to the manufacturer instructions. This is a colorimetric detection and quantification method which utilizes bicinchoninic acid (BCA) as the detection reagent for Cu1+ formed when Cu2+ is reduced by the protein in an alkaline environment. Quadruplicates of lysed cells per culturing time were incubated at 37 °C for 2 h. A purple-colored reaction product was measured at 562 nm in a microplate reader (SynergieHT, Bio-Tek, USA) and calculated based on an albumin standard curve, ranging from 0 to 40 μg mL−1.
RNA isolation and real-time quantitative polymerase chain reaction
At each culturing time, the hBMSCs were washed with PBS, immersed in Tri® reagent (Sigma-Aldrich, USA) and stored at −80 °C until further use. Proteins were removed with chloroform; isoamylalkohol (BioChemica, AppliChem, Germany) extraction and the RNA pellets were washed once with 2-propanol (Sigma-Aldrich; USA) and once with 70% ethanol (Panreac, Spain). The total RNA pellets were reconstituted in Rnase free water (Gibco, Invitrogen, UK). Determination of the RNA concentration for each replica (quadruplicates of each condition per time point) was performed by microspectrophotometry (NanoDrop 1000, Thermo Scientific, USA).Reverse transcriptase (RT)-PCR was performed according to the protocol from iScript™ cDNA synthesis kit (Quanta BioSciences™, USA). Briefly, a reaction mixture consisting of 1X iScript Reaction Mix, 1 μL iScript Reverse Transcriptase, 100–150 ng RNA template and nuclease-free water was prepared, in 20 μL of total volume. The single-strand cDNA synthesis occurred by incubating the complete reaction mixture 5 min at 22 °C, followed by 30 min at 42 °C and terminated by an incubation at 85 °C for 5 min.
Amplification of the target cDNA for real-time PCR quantification were performed according to the manufacturer instructions, using 2 μL RT cDNA products, 250 nM each primer 1X PerfeCta® SYBR® Green FasterMix® (Quanta BioSciences™, USA) and nuclease-free water, in a final volume of 25 μL. Forty-four cycles of denaturation (95 °C, 10 s), annealing (temperature dependent on the gene, 30 s) and extension (72 °C, 30 s) were carried out in the Mastercycler® epgradient S realplex thermocycler (Eppendorf, Germany) for all genes. The transcripts expression data were normalized to the housekeeping gene glyceraldehydes-3-phosphate-dehygrogenase (GAPDH) and the quantification performed according to the Livak method (2−ΔΔCt method), considering the Basal culture condition as calibrator.
Statistical analysis
The data was statistically analyzed using IBM SPSS software (version 20; SPSS Inc.). We first applied the Shapiro–Wilk test to test the assumption of normality and the results showed that the data was not following a normal distribution. Consequently, nonparametric way of Kruskal Wallis test, followed by Dunnett's test for multiple comparisons to Basal and followed by Dunnett's test for multiple comparisons to osteogenic. P values lower than 0.01 were considered statistically significant in the analysis of the results.Results
Development and characterization of Dex–AscA-loaded liposomes
In this work, we prepared six different liposomal formulations, including three conventional liposome (CL) formulations A, B and C, and three sterically stabilized liposome (SSL) with DSPE–PEG (formulation D), and SSL with DSPE–PEG and PE–Rho (formulation E and F). The CL and SSL differ in the presence of a polymer coating at the surface of the liposome, in this case polyethylene glycol (PEG). In the previous study, a maximum Dex encapsulation was achieved with the Dex:HSPC ratio of 0.25:2.48 In this work, Dex:HSPC was kept constant and the concentration of AscA was varied from 10 to 50 mg. The PL and EE are presented in Table 2. The PL of AscA significantly increased from 0.77 ± 18.04 to 1.80 ± 20.98 (mol mol−1), whereas the EE of AscA decreased from 15.44 ± 8.91 to 7.20 ± 1.79% when AscA was increased from 10 to 50 mg. The PL results show that 180 molecules of AscA are encapsulated in the inner core, and 43 molecules of Dex are incorporated within the bilayer per 100 molecules of lipids. With the data obtained in this study a compromise was obtained with the formulation 0.25:2 (Dex:HSPC) and 50 mg of AscA, that was subsequently used in the Dex–AscA release study.
| AscA (mg) | Dex:HSPC |
PL (mol mol−1) | EE (%) | |||
|---|---|---|---|---|---|---|
| AscA | Dex | AscA | Dex | |||
| A | 10 | 0.25:2 |
0.77 ± 18.04 | 0.46 ± 0.13 | 15.44 ± 8.91 | 28.79 ± 6.95 |
| B | 25 | 0.25:2 |
0.91 ± 30.94 | 0.50 ± 0.09 | 7.31 ± 4.16 | 32.13 ± 13.78 |
| C | 50 | 0.25:2 |
1.80 ± 20.98 | 0.43 ± 0.11 | 7.20 ± 1.79 | 23.79 ± 6.37 |
Table 3 presents the size and ζ-potential of the liposomes formulations. The ζ-potential of the conventional liposomes (formulations A, B and C) was close to neutrality. However, the ζ-potential increased for the PEGylated liposomes (formulations D, E and F). The particle size distribution of the liposomes prepared in this study showed a monodisperse distribution.
| Formulation | ζ-Potential | Size (nm) |
|---|---|---|
| A | 0.63 ± 1.76 | 152.16 ± 2.09 |
| B | −1.19 ± 0.60 | 153.90 ± 2.80 |
| C | −0.50 ± 0.48 | 133.70 ± 1.79 |
| D | −15.50 ± 0.70 | 143.60 ± 4.94 |
| E | −21.60 ± 1.10 | 115.70 ± 5.04 |
| F | −18.03 ± 0.56 | 124.00 ± 2.85 |
Dex and AscA release from liposomes
The release profile of Dex and AscA from the PEGylated liposomes was performed during 21 days, using the liposomal formulation D. This timeframe was selected in accordance with the culture time usually required to obtain a complete osteogenic differentiation of MSCs in vitro.The release profiles of Dex and AscA are presented in Fig. 1, and that data was fitted with mathematical models in order to study the Dex and AscA release mechanisms (Table 4). Dex was released more rapidly from the liposomes when compared to AscA. The Dex release profile showed an initial burst release within 12 h. Afterwards, a slower but sustained release was observed until 21 days. Based on the non-linear regression of Dex release, eqn (3) was fitted to the release data achieving an R2 = 0.96 and a release exponent of n = 0.24. Inversely, AscA was released without an initial burst. Indeed, the release of AscA from the liposomes was not detected until 6 h. However, AscA was slowly released from 24 h until day 21. Therefore, AscA is released in a linear fashion after 24 h (Adj R2 = 0.99).
 | ||
| Fig. 1 In vitro cumulative release of Dex and AscA from liposomes (formulation E). The experiments were performed in triplicate. | ||
| AscA | Dex | |||||
|---|---|---|---|---|---|---|
| Linear | Power law | |||||
| a | k | Adj R2 | a0 | k (day−n) | n | Adj R2 |
| 0.33 | 0.021 | 0.99 | −0.17 | 0.23 | 0.24 | 0.96 |
Biological activity
Cell viability, proliferation and protein assessment
The influence of a single dose and repeated doses of Dex-loaded liposomes (E) and Dex–AscA-loaded liposomes (F) in basal (Basal, E(once), E(repeat), F(once), F(repeat)), and osteogenic medium (Osteo, E(once), E(repeat), F(once), F(repeat)) over hBMSCs viability and proliferation was assessed (Fig. 2). | ||
| Fig. 2 Box plot of hBMSCs metabolic activity ((Abs 490 nm) per 106 cells) (A and B), proliferation (DNA conc. (μg mL−1)) (C and D), and protein concentration (protein conc. (ng per mL) per 106 cell) (E and F) when cultured in basal medium (Basal, E(once), E(repeat), F(once), F(repeat)), and osteogenic medium (Osteo, E(once), E(repeat), F(once), F(repeat)) after 7, 14 and 21 days of culture. The experiments were performed three times independently, with sample triplicates. a denotes significant differences compared to Basal and b denotes significant differences compared to Osteo. | ||
In terms of cell viability/cell number, at the 7th, 14th and 21st day, no significant differences were found between culture conditions and the negative control Basal (Fig. 2A). Comparing with the osteogenic medium conditions (Fig. 2B), at 7th and 14th day, culture conditions E(repeat) and Osteo displayed significantly higher cell viability than the negative control condition Basal (p < 0.01). When using the culture condition Osteo as positive control (Fig. 2B), at the 14th day, the culture conditions E(repeat), and F(once) displayed significantly higher cell viability (p < 0.001). At the 7th and the 21st days, no significant differences were found between all the culture conditions and the control condition Osteo. Comparing with the basal medium conditions (Fig. 2A), at the 14th day, culture conditions Basal and F(repeat) displayed significantly higher cell viability than the control condition Osteo (p < 0.001).
In terms of cell proliferation (Fig. 2C) and considering the negative control condition Basal, at the 7th day, F(repeat) displayed significantly lower cell proliferation (p < 0.001). At the 14th and 21st day, no significant differences were found between all the culture conditions and the negative control Basal. Comparing with the osteogenic medium conditions (Fig. 2D), at the 7th day, the culture conditions E(once), F(once) and F(repeat), and at the 14th day, the culture conditions E(once) and F(once) displayed significantly lower cell proliferation than the negative control condition Basal (p < 0.001). When using the culture condition Osteo as positive control (Fig. 2D), at the 7th and 14th days, repeated doses of Dex-loaded liposomes, E(repeat), displayed significantly lower cell proliferation (p < 0.01). At the 21st day of hBMSCs culture, no significant differences were found between all the culture conditions and the positive control Osteo.
In terms of protein synthesis (Fig. 2E) and considering the negative control condition Basal, at 7th and 14th day, no significant differences were found. However, at the 21st day, the cultures conditions E(once), E(repeat), and F(repeat) displayed significantly lower protein concentration than the negative control Basal (p < 0.001). Comparing with the osteogenic medium conditions (Fig. 2F), at the 7th day, the culture condition Osteo, and at the 21st day, the culture conditions Osteo, and F(repeat) displayed significantly lower protein concentration than the negative control Basal (p < 0.001). When using the culture condition Osteo as positive control (Fig. 2F), at the 7th, 14th, and 21st day, no significant differences were found between conditions. Comparing with the basal medium conditions (Fig. 2E), at the 7th day, the culture conditions Basal and F(once), and at the 21st day, the culture condition Basal displayed significantly higher protein concentration than the positive control Osteo (p < 0.001).
Alkaline phosphatase activity
To assess the onset of the osteoblastic activity of the cultured hBMSCs, the quantification of the alkaline phosphatase (ALP) enzyme activity was performed according to the p-nitrophenol assay. Considering the culture condition Basal as negative control, at the 7th day, only the culture condition F(repeat) displayed significantly higher ALP activity (p < 0.001) (Fig. 3A). At the 14th and the 21st days, the culture conditions E(repeat) and F(repeat) displayed significantly higher ALP activity than the negative control condition Basal (p < 0.001). Comparing with the osteogenic medium condition (Fig. 3B), at the 7th day, the culture condition F(repeat), at the 14th and the 21st days, the culture conditions Osteo, E(repeat), and F(repeat) displayed significantly higher ALP activity than the negative control condition Basal (p < 0.001).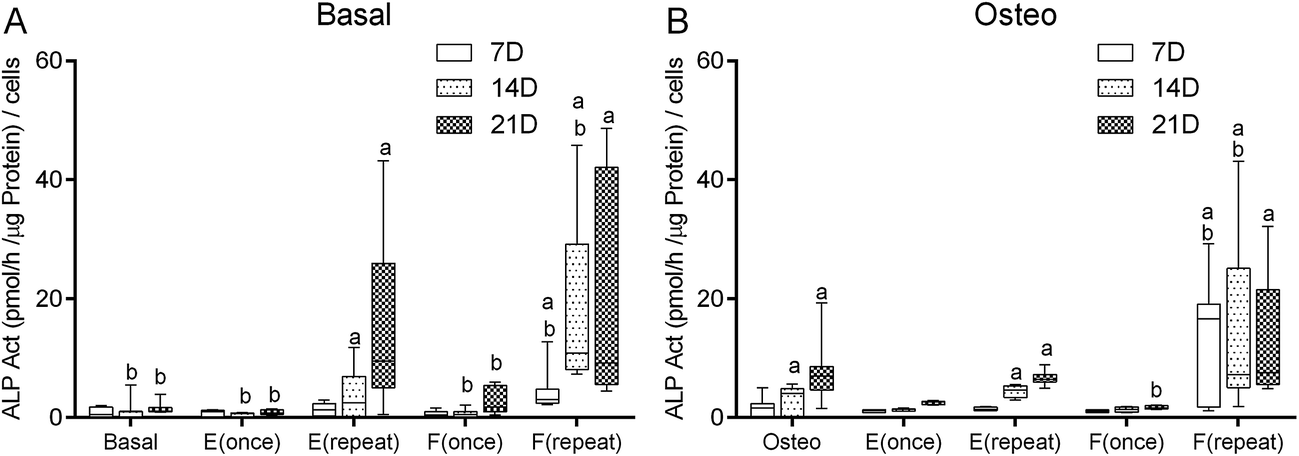 | ||
| Fig. 3 Box plot of hBMSCs ALP activity (pmol per h per μg per protein per cells) when cultured in basal medium (Basal, E(once), E(repeat), F(once), F(repeat)) (A), and osteogenic medium (Osteo, E(once), E(repeat), F(once), F(repeat)) (B) after 7, 14 and 21 days of culture. The experiments were performed three times independently, with sample triplicates. a denotes significant differences compared to Basal and b denotes significant differences compared to Osteo. | ||
When considering the culture condition Osteo as a positive control (Fig. 3B), at 7th and 14th day, the culture condition F(repeat) displayed significantly higher ALP activity (p < 0.001). At the 21st day, the culture condition F(once) displayed significantly lower ALP activity than the positive control condition Osteo (p < 0.001). Comparing with the basal medium conditions (Fig. 3A), at 7th and 14th day, the culture condition F(repeat) displayed significantly higher ALP activity than the positive control condition Osteo (p < 0.001). Inversely, at the 14th day, the culture conditions Basal, E(once) and F(once) displayed significantly lower ALP activity than the positive control condition Osteo (p < 0.001). Also, at the 21st day, the culture conditions Basal, E(once), and F(once) displayed significantly lower ALP activity than the positive control condition Osteo (p < 0.001).
Genotypic expression of osteoblastic markers
The influence of a single dose and repeated doses of Dex-loaded liposomes (E) and Dex–AscA-loaded liposomes (F), in basal and osteogenic medium over hBMSCs genotypic expression was assessed. Changes over time of specific osteoblastic differentiation markers are presented in bar plots for the different conditions (Fig. 4 and 5). | ||
| Fig. 4 Bar plot of hBMSCs ALP, Runx2, OP, OCN, Osterix, BSP and Col Ia genes expression cultured in basal medium (Basal, E(once), E(repeat), F(once), F(repeat)), and osteogenic medium (Osteo, E(once), E(repeat), F(once), F(repeat)) after 7, 14 and 21 days of culture. The experiments were performed three times independently, with sample triplicates. a denotes significant differences compared to Basal and b denotes significant differences compared to Osteo. | ||
 | ||
| Fig. 5 Line plot of hBMSCs ALP, Runx2, OP, OCN, Osterix, BSP and Col Ia genes expression cultured in basal medium (Basal, E(once), E(repeat), F(once), F(repeat)), and osteogenic medium (Osteo, E(once), E(repeat), F(once), F(repeat)) after 7, 14 and 21 days of culture. The experiments were performed three times independently, with sample triplicates. | ||
In terms of ALP gene expression, and considering the basal medium conditions (Fig. 4A) and the negative control condition Basal, at the 7th and 14th day, the conditions E(repeat), F(once) and F(repeat) displayed significantly higher ALP (p < 0.001). At the 21st day, E(repeat), and F(repeat) displayed significantly higher ALP than the negative control Basal. Comparing with the osteogenic medium conditions (Fig. 4B), at the 7th and 14th day, the conditions Osteo, E(repeat), and F(repeat) displayed significantly higher ALP than the negative control Basal (p < 0.001). At the 21st day, the culture conditions Osteo, E(repeat), and F(repeat) displayed significantly higher ALP than the negative control Basal. Considering the culture condition Osteo as positive control (Fig. 4B), at the 7th, 14th and 21st day, no significant differences were found between the conditions. Comparing with the basal medium conditions (Fig. 4A), at 7th day no significant differences were found. However, at the 14th day, only the culture condition Basal displayed significantly lower ALP expression than the positive control condition Osteo (p < 0.01). At the 21st day, the culture conditions Basal, E(once) and F(once) displayed significantly lower ALP gene expression than the positive control condition Osteo (p < 0.01). These observations at the ALP gene expression are in accordance with the results achieved in the ALP activity quantification.
In terms of Runx2 expression, and considering the basal medium conditions (Fig. 4C) and the negative control condition Basal, at the 7th, 14th and 21st days, no significant differences were found between the culture conditions. Comparing with the osteogenic medium conditions (Fig. 4D), at the 14th day, F(repeat) displayed significantly higher Runx2 levels than the negative control condition Basal (p < 0.01). At the 21st day, the culture condition Osteo displayed significantly higher Runx2 expression than the negative control condition Basal (p < 0.01). Considering the culture condition Osteo as a positive control (Fig. 4D), at the 7th, 14th and 21st days, no significant differences were found between all culture conditions in terms of Runx2 expression. Comparing with the basal medium conditions (Fig. 4C), at the 21st day, only the condition Basal displayed significantly lower Runx2 expression than the positive control condition Osteo (p < 0.01).
In terms of osteopontin (OP) expression, and considering the basal medium conditions (Fig. 4E) and the negative control condition Basal, at the 7th, 14th and 21st days, no significant differences were found between the culture conditions. Considering the culture condition Osteo as a positive control (Fig. 4F), at the 21st day; no significant differences were found between culture conditions. However, at the 7th and 14th day, F(once) displayed significantly higher OP than the positive control condition Osteo (p < 0.01).
In terms of osterix (Fig. 4G and H), osteocalcin (OCN, Fig. 4I and J) and bone sialoprotein (BSP, Fig. 4K and L) expression at the 7th, 14th and 21 days, no significant differences were observed between all culture conditions and the controls Basal and Osteo. In terms of collagen type 1 alpha (Col Ia) expression (Fig. 4M), at the 7th, 14th and 21st days, no significant differences were found between conditions and the negative control Basal. Comparing with the osteogenic medium conditions (Fig. 4N), at the 7th day, the culture condition Osteo displayed significantly lower Col Ia than the negative control Basal (p < 0.01). Considering the culture condition Osteo as a positive control (Fig. 4N), at the 7th day, E(repeat) displayed significantly higher Col Ia (p < 0.01). At the 14th day, no significant differences were found between all culture conditions and the positive control Osteo; at the 21st day, the culture condition F(repeat) also displayed significantly lower Col Ia than the positive control condition Osteo (p < 0.01). Comparing with the basal medium conditions (Fig. 4M), at the 7th day, the culture condition Basal, and at the 21st day, the culture condition E(repeat) displayed significantly lower Col Ia than the positive control condition Osteo (p < 0.01).
Comparing the different time points (Fig. 5) we observed that, in terms of ALP gene expression, F(once) in basal medium at 7th day displayed a significantly higher gene expression than 14th and 21st days (p < 0.05). No significant differences between the days was found in the other culture conditions. In terms of Runx2 gene expression, Osteo at 21st day displayed a significantly higher gene expression than 7th and 14th days (p < 0.05). In terms of OP gene expression, F(once) in osteogenic medium at 7th day displayed a significantly higher gene expression than the 21st day (p < 0.05). In terms of Osterix gene expression, E(repeat) in basal medium at 21st day displayed a significantly higher gene expression than the 7th day (p < 0.05). In terms of OCN gene expression, E(repeat) in basal medium at 14th day displayed a significantly higher gene expression than the 7th day (p < 0.05). In terms of BSP gene expression, E(repeat) in basal medium at 21st day displayed a significantly higher gene expression than the 7th and 14th day (p < 0.05). In terms of Col Ia gene expression, Osteo condition at 21st day displayed a significantly higher gene expression than the 7th and 14th day (p < 0.05).
Discussion
Development and characterization of Dex–AscA-loaded liposomes
We encapsulated Dex and AscA into liposomes because they are usual supplements used as differentiation factors involved in the in vitro osteogenic differentiation of MSCs.41,52 Encapsulation of Dex and AscA into liposomes can possibly give the following benefits to the culture of MSCs:52–54 (i) protection from oxygen, moisture, temperature and light, which results in increased stability during handling and storage; (ii) protection from undesirable reactions between one specific bioactive agent and others present in the culture medium; (iii) increases the solubility, diffusivity and bio-distribution; (iv) decreases toxicity; (v) improved bioavailability, sustained delivery efficacy and avoid side effects; (vi) possibility to deliver AscA and Dex inside the cells.AscA encapsulation can be difficult because of its water solubility and reactivity.55 The stability of AscA is strongly dependent on the pH and temperature.56 The rate of oxidation increases at pH values close to its pKa1.57 Therefore, acidification is frequently used to stabilize the AscA. The half-life of AscA in drinking water and phosphate buffers at neutral pH is less than 1 h.58 Therefore, the incorporation of AscA into liposomes is one possible strategy to ensure AscA stabilization.
Our results showed that the liposomes encapsulated more AscA than Dex (Table 2). This is explained by the fact that there is more space in the inner compartment to encapsulate hydrophilic molecules than within the lipid bilayer to encapsulated lipophilic molecules. The thickness of the lipid bilayer may vary accordingly to the type of lipids used to form the liposomes.59 Tsotas et al.60 compared Dex-loaded liposomes prepared with PC and DSPC. In plain lipid vesicles, the encapsulation efficiency of Dex was higher for the DSPC liposomes than to the PC liposomes. This effect was attributed to the longer acyl chain length of DSPC (C18) which may provide more space within the bilayer for the incorporation of Dex molecules.60 AscA was encapsulated into DPPC and DPPC/cholesterol liposomes.56 It was demonstrated that co-encapsulation of citric and ascorbic acids into liposomes results in considerable stabilization of the AscA in model systems containing catalytic concentrations of copper ions. The rate of AscA oxidation was decreased by up to 300-fold than in free AscA. Also, the incorporation of cholesterol into the membranes resulted in lower stabilization efficacy at room temperature and better thermal stability above the phase temperature of DPPC liposomes (Tc = 41 °C).56 In this work, we used HSPC which has a high phase transition temperature, i.e. Tc = 52 °C,61 and cholesterol was not used because it impairs the encapsulation of Dex.
The relatively low degree of encapsulation of AscA could be ascribed to the low phospholipid/buffer ratio or to the encapsulation method used to prepare the Dex–AscA-liposomes. In the lipid film method, the presence of large amounts of external aqueous medium induces a low encapsulation of hydrophobic drug.62 Higher degrees of encapsulation could be obtained by freeze drying, rehydration/dehydration cycles or by direct injection method.25 We could notice that increasing the concentration of AscA in the buffer medium impairs the formation of the liposomes. This may be related with the pH of the buffer solution: while increasing the AscA concentration, the pH of the buffer decreases, provoking hydrolysis of the liposomes.63 It was demonstrated that hydrolysis of liposomes at low pH, results in the formation of lysophospholipids and free fatty acids that can destabilize the liposomes.63,64 Hydrolysis can significantly increase the permeability of bilayers that are in the solid-ordered gel phase.64 One could increase the pH, however, high pH affects the stability of the AscA.56
The prepared liposomes showed a monodisperse size distribution and the ζ-potential increased for the PEGylated liposomes (formulations D and E, Table 3). The ζ-potential of the liposomes is negative due to the presence of the terminal carboxylic groups in the lipids. This negative charge may reduce the electrophoretic mobility of liposomes and create a high-energy barrier that avoids agglomeration and stabilizes the nanosuspension.48,65
The release of bioactive agent from the liposomes is determinant in its biological effect; thus the evaluation of the release kinetics is of paramount importance. The use of a kinetic model is often helpful to elucidate the release mechanisms, control of bioactive agent release and also to optimize the loading process.66 Drug release profiles from liposomes described in the literature characteristically show an initial fast drug release (usually referred to as a burst) followed by a slower but steady release.67 The initial fast rate of release is commonly ascribed to drug detachment from the liposomal surface while the later one results from the sustained drug release from the inner compartment.67 Water is released from the liposomes through pores formed in their membrane.59 Our release studies showed that the release profile of Dex from the liposomes was biphasic, showing a relatively large burst effect over the first 12 hours, followed by a slower release phase (Fig. 1). The Dex release data was fitted with a non-linear function using the equation eqn (3), and the coefficient of determination (R2) was also determined. The objective behind fitting this equation to the bioactive agent release data was to understand the underlying phenomena involved in the drug release mechanism. The parameters a0, and k define the burst stage and the bigger values they have, the higher extent of burst. On the other hand, n indicates the magnitudes of the drug desorption/diffusion stage and the higher values they have, more sustained is the release. The R2 was of 0.96, which indicates that the data was well fitted by the model. However, the value of the release exponent (n = 0.24) is beyond the limits of Korsmeyer model.50 This means that the Dex released from the liposome was controlled by the diffusion drug release mechanism. Similar release profile of Dex was observed in previous studies.21,24,48,60 Although, Dex release kinetics were similar for different liposome types, and it was concluded that the drug release is due mainly to dilution of liposome dispersions that leads to repartitioning of Dex.60 Also the incorporation of Dex within the liposome bilayer may influence the liposomes' stability.21 It may interact differently with phospholipid acyl chains and head groups destabilizing the membrane. Kawakami et al. reported the modeling of release kinetics of poorly water-soluble drug molecules from liposomes.68 The kinetic rate equations take in account two transport mechanisms: collisions between liposomes and drug diffusion through the aqueous phase. The first mechanism lead to an apparent first-order kinetic behavior, and in the second mechanism, the behavior may become bi-exponential or sigmoidal.68
With a distinct behavior, we observed a slower release of AscA from the liposomes, where it was possible to fit a linear regression. The R2 was of 0.99, which indicates that the data was well fitted by the zero order release model. This indicates a process of constant drug release from a liposome. This slower release may be related to the high transition temperature of HSPC. Indeed, at physiological conditions the liposomes are in a gel phase. Furthermore, the difference on the release profile of both bioactive agents may be related with their localization within the liposomes. Dex is encapsulated within the bilayer compartment and may be faster released than the AscA, which is encapsulated in the inner core compartment. Therefore, being AscA a hydrophilic molecule, it is difficult to pass through the liposome' hydrophobic compartment.
Dex and AscA have to be delivered at the right concentration and at the right time in order to induce the osteogenic differentiation of the MSCs. The concentration of Dex released is in accordance with the concentration of Dex presented in the standard osteogenic culture medium. The concentration of AscA released is lower than concentration of AscA presented in the standard osteogenic culture medium. As was discussed above, the liposomes encapsulated more AscA than Dex. Therefore, part of the AscA remained into the liposomes. It is well described in the literature the effects of osteogenic differentiation factors on intracellular signaling cascades that lead to osteogenic differentiation of hBMSCs.69 Dex induces runt-related transcription factor-2 (Runx2) expression by FHL2/beta-catenin-mediated transcriptional activation and that Dex enhances Runx2 activity by upregulation of TAZ and MKP1.69 AscA leads to the increased secretion of collagen type I (Col1), which in turn leads to increased Col1/alpha2beta1 integrin-mediated intracellular signaling.69 Thus, firstly Dex has to be delivered to activate Runx2, then, AscA has to be delivered to activate Col1.
Biological activity of Dex–AscA released from liposomes
The precise cellular mechanisms by which bone synthesis is controlled within developing progenitor cells is unclear.70 The possibility to culture and induce the differentiation of human MSCs provide a helpful model for evaluating the multiple factors responsible for the step-wise progression of cells from undifferentiated precursors to secretory osteoblasts.41 Therefore, optimal combination of growth differentiation factors, cytokines and serum supplements, and their concentration within the media is essential for their application in modern tissue engineering.32Herein, the effect of Dex-loaded liposomes and Dex–AscA-loaded liposomes (single dose or repeated doses, in basal and osteogenic medium) on viability, proliferation, protein synthesis and osteogenic differentiation of hBMSCs was assessed. In this study, we did not focus on the uptake of the liposomes by hBMSCs in the different culture conditions. However, it is well documented that the nanoscale properties of the nanoparticles determine the solubility, diffusivity, bio-distribution, biological fate, toxicity, site specific ligands to achieve active targeting, and increase the efficacy and therapeutic index of bioactive agents.2,17 It is also reported that nanoparticles with 100 nm in diameter is an optimum size for cell internalization.73,74 Moreover, the cellular internalization behavior differs from cell types, incubation time, culture medium, nanoparticle concentration and properties such as composition and surface charge.2,17,75 Kettler et al.75 showed that uptake kinetics of silver nanoparticles into pulmonary epithelial cells was highly influenced by medium composition. Uptake into cells was higher in medium without foetal calf serum (FCS), reaching approximately twice the concentration after 24 h than in medium supplemented with FCS.75 Our study also shows that the culture medium does effect biological results, and consequently, may have an effect on uptake of the liposomes by the hBMSCs.
Cell viability data showed that Dex-loaded liposomes and Dex–AscA-loaded liposomes are not cytotoxic to hBMSCs (Fig. 2). In basal conditions, the proliferation of hBMSCs was not significantly affected by the supplementation of medium by Dex-loaded or Dex–AscA-loaded liposomes, in single or repeated doses (Fig. 2). But, the cell proliferation decreased when hBMSCs were cultured in osteogenic medium with E(once), E(repeat) and F(once) (Fig. 2). Nevertheless, similar proliferation levels to the controls (Basal and Osteo) were observed when the hBMSCs were cultured in osteogenic medium (just with β-GP) supplemented with Dex–AscA-loaded liposomes (i.e. F(repeat)). This result can be explained by the fact that both Dex and AscA were encapsulated into the liposomes, and their concentration was sufficient to stimulate the proliferation of the hBMSCs. Choi et al.52 studied the effect of AscA on bone marrow-derived mesenchymal stem cell proliferation and differentiation in the culture medium. AscA stimulates the proliferation of MSC in vitro. At specific concentrations, AscA acts as a growth promoter to increase MSCs proliferation and DNA synthesis of cells in the culture medium.52 However, high concentrations of AscA are cytotoxic and can lead to inhibition of cell proliferation and consequent apoptosis. Moreover, the cytotoxicity of AscA is related to the culture condition factors such as the type of medium or incubator condition (i.e. CO2 concentration).52 Moreover, when MSCs are cultured in the presence of AscA, the effects of Dex are masked.32 Our results showed that, Dex encapsulated within the lipid bilayer of liposomes inhibited the hBMSCs proliferation when AscA and β-GP were added in the culture medium, in single or repeated doses (i.e. E(once) and E(repeat) in Osteo medium, Fig. 2D). The effect of Dex on cell proliferation has been shown to depend on the concentration and the osteoprogenitor maturity.41,42 Dex acts to direct osteoprogenitor cells from a proliferation state to a matrix maturation stage.42 Altogether, these results indicate that the concomitant encapsulation of Dex and AscA into the liposomes promote the proliferation of hBMSCs preferentially in basal medium.
In basal conditions, the protein synthesis of hBMSCs was not significantly affected by the supplementation of medium by Dex-loaded or Dex–AscA-loaded liposomes, in single or repeated doses at the 7th and 14th day (Fig. 2E and D). However, at the 21st day, the protein synthesis significantly decreased for E(once), E(repeat), F(repeat) in basal medium and F(repeat) in osteogenic medium conditions. This result can be explained by the differentiation of the hBMSCs into the osteogenic lineage. In osteogenic medium conditions, the protein synthesis of hBMSCs was not significantly affected by the supplementation of medium by Dex-loaded or Dex–AscA-loaded liposomes, in single or repeated doses (Fig. 2D).
Alkaline phosphatase activity is one of the most used indicators of the onset of MSCs osteoblastic differentiation in vitro.71 Osteoblastic differentiation in vitro is marked by three distinct stages of cellular activity: proliferation, extracellular matrix (ECM) maturation and matrix mineralization.72 When MSCs are induced to differentiate into osteoblasts, MSCs tend to change from a fibroblastic morphology to a cuboidal shape, starting to produce a mature ECM mainly composed of collagen type I (Col Ia). During this stage, the rate of proliferation decreases and the expression of ALP increases.72 Following this period of matrix maturation, cellular aggregates or nodules begin to mineralize the ECM.32 Therefore, the expression of osteocalcin (OCN) increases simultaneously to the mineral deposition while ALP levels decline.72 Our results point out that hBMSCs cultured in the presence of Dex-loaded liposomes or Dex–AscA-loaded liposomes given in a single dose, in basal or osteogenic medium, did not significantly increased the level of ALP activity relatively to the negative control (Basal) or the positive control (Osteo) (Fig. 3). This means that a single dose of growth/differentiation factors-loaded liposomes in both culture media was not sufficient to induce the osteogenic differentiation of hBMSCs. This observation can be explained by the low concentration of Dex released from the liposomes into the culture medium along the entire timeframe of the experiment. In the literature it has been suggested that the optimal osteogenic differentiation is achieved at a Dex concentration of 100 nM.41 Although a single dose of Dex- or Dex–AscA-loaded liposomes released the same concentration of Dex presented in the standard osteogenic culture medium, this concentration is no longer available after the first medium change at the 3rd day of culture. Conversely, repeated doses of Dex-loaded or Dex–AscA-loaded liposomes in both culture media significantly increased the ALP activity, comparing to the control conditions (i.e. Basal and Osteo). Indeed, this dose dependent effect of Dex has been previously reported in the literature.41,42 Encapsulation of AscA into the liposomes had a positive effect on the differentiation of hBMSCs. Repeated doses of Dex–AscA-loaded liposomes, in basal and incomplete osteogenic medium, increased the level of ALP activity (Fig. 5). The differentiation of MSC also depends on ascorbic acid concentration.52 It is known that AscA regulates cell differentiation, particularly the multilineage differentiation capacity of mesenchymal stem cells.52 AscA acts as a co-factor in the post-translational modification of collagen molecules and, therefore, increases collagen production.52 These results also indicate that the dual release of Dex and AscA from the liposomes successfully promotes the differentiation of hBMSCs in basal medium.
It is important to highlight that repeated doses of Dex-loaded and Dex–AscA-loaded liposomes in basal medium increased the ALP activity of the hBMSC when compared to the complete osteogenic medium (Osteo). This observation means that it is not needed to use all the differentiation factors (i.e. Dex, AscA and β-GP) in the culture medium to induce the in vitro differentiation of the hBMSCs. Indeed, this hypothesis was previously demonstrated when hBMSCs were culture onto electrospun nanofiber meshes with Dex-release capability.36 To corroborate this hypothesis, the genotypic expression pattern of the osteogenic markers was performed (Fig. 4). It is supposed that Dex acts through the modulation of nuclear steroid receptors, inducing transcriptional effects which lead to the activation of specific pathways converging to bone formation.32,42 Runx2 affects the expression of bone-specific genes such as Osterix, Col Ia, OCN and BSP, by binding to the promoters of these genes. Generally, Runx2, ALP and Col Ia are known to be early markers of osteoblastic differentiation, whereas OCN and osteopontin (OP) are expressed later in the differentiation process.32 Our osteoblastic gene expression data showed that repeated doses of Dex-loaded and Dex–AscA-loaded liposomes only increased the expression of ALP gene compared to the control (Osteo). All the other osteogenic markers were expressed in all culture conditions, meaning that the hBMSCs were induced to differentiate into the osteogenic lineage.
Conclusion
In this work, we showed the versatility of liposomes to encapsulate two osteogenic differentiation factors, their kinetics of release, and their effect over hBMSCs. Dex and AscA were successfully encapsulated into the liposomes. The release study showed a burst release of Dex and a slow but sustained release of AscA. The biological results indicate that Dex–AscA-loaded liposomes do not have any cytotoxicity and were able to promote the induction of the differentiation of hBMSCs into the osteogenic lineage in both basal and osteogenic medium. This dual release system strategy for the induction of the differentiation of hBMSCs can be transposed to other tissue engineering and regenerative approaches, namely on the implantation of biofunctionalized scaffolds with surface-immobilized Dex–AscA-loaded liposomes. Alternatively, the liposomes can be directly injected at the injury site to induce the differentiation of local or recruited endogenous MSCs in vivo.Acknowledgements
The authors thank the Portuguese Foundation for Science and Technology for the PhD grant of N. S. Monteiro (SFRH/BD/62465/2009), the Investigator FCT starting grant of A. Martins (IF/00376/2014). This work was supported by OsteoGraphy (PTDC/EME-MFE/2008) and MaxBone (PTDC/SAU-ENB/115179/2009) projects. It was also partly supported by the POLARIS (FP7-REGPOT-2012-2013-1) and RL3 – TECT – NORTE-01-0124-FEDER-000020, co-financed by North Portugal Regional Operational Programme (ON.2 – O Novo Norte), under the National Strategic Reference Framework (NSRF), through the European Regional Development Fund (ERDF).References
- V. E. Santo, M. E. Gomes, J. F. Mano and R. L. Reis, Tissue Eng., Part B, 2012, 19(4), 327–352 CrossRef PubMed.
- N. Monteiro, A. Martins, R. L. Reis and N. M. Neves, Regenerative Therapy, 2015, 1, 109–118 CrossRef.
- T. P. Richardson, M. C. Peters, A. B. Ennett and D. J. Mooney, Nat. Biotechnol., 2001, 19, 1029–1034 CrossRef CAS PubMed.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- C. A. Simmons, E. Alsberg, S. Hsiong, W. J. Kim and D. J. Mooney, Bone, 2004, 35, 562–569 CrossRef CAS PubMed.
- J. S. Park, H. N. Yang, D. G. Woo, S. Y. Jeon and K. H. Park, Biomaterials, 2011, 32, 28–38 CrossRef CAS PubMed.
- M. S. Bhojani, M. Van Dort, A. Rehemtulla and B. D. Ross, Mol. Pharm., 2010, 1921–1929, DOI:10.1021/mp100298r.
- N. Bertrand and J.-C. Leroux, J. Controlled Release, 2012, 161, 152–163 CrossRef CAS PubMed.
- Z. Skeete, H. Cheng, E. Crew, L. Lin, W. Zhao, P. Joseph, S. Shan, H. Cronk, J. Luo, Y. Li, Q. Zhang and C.-J. Zhong, ACS Appl. Mater. Interfaces, 2014, 6, 21752–21768 CAS.
- V. E. Santo, M. E. Gomes, J. F. Mano and R. L. Reis, Nanomedicine, 2012, 7, 1045–1066 CrossRef CAS PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- M. Chorny, I. Fishbein, H. D. Danenberg and G. Golomb, J. Controlled Release, 2002, 83, 389–400 CrossRef CAS PubMed.
- M. Hasan, G. Ben Messaoud, F. Michaux, A. Tamayol, C. J. F. Kahn, N. Belhaj, M. Linder and E. Arab-Tehrany, RSC Adv., 2016, 6, 45290–45304 RSC.
- D. Y. Hegh, S. M. Mackay and E. W. Tan, RSC Adv., 2016, 6, S6859–S6866 RSC.
- M. Kulkarni, U. Greiser, T. O'Brien and A. Pandit, Trends Biotechnol., 2010, 28, 28–36 CrossRef CAS PubMed.
- P. Tayalia and D. J. Mooney, Adv. Mater., 2009, 21, 3269–3285 CrossRef CAS PubMed.
- N. Monteiro, A. Martins, R. L. Reis and N. M. Neves, J. R. Soc., Interface, 2014, 11, 20140459 CrossRef.
- A. K. Jain, A. Agarwal, H. Agrawal and G. P. Agrawal, J. Liposome Res., 2012, 22, 205–214 CrossRef CAS PubMed.
- L. C. Gomes-da-Silva, N. A. Fonseca, V. Moura, M. C. P. de Lima, S. Simoes and J. N. Moreira, Acc. Chem. Res., 2012, 45, 1163–1171 CrossRef CAS PubMed.
- J. S. Kauffman, B. M. Ellerbrock, K. A. Stevens, P. J. Brown, W. T. Pennington and T. W. Hanks, ACS Appl. Mater. Interfaces, 2009, 1, 1287–1291 CAS.
- U. Bhardwaj and D. J. Burgess, Int. J. Pharm., 2010, 388, 181–189 CrossRef CAS PubMed.
- N. Monteiro, D. Ribeiro, A. Martins, S. Faria, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, ACS Nano, 2014, 8, 8082–8094 CrossRef CAS PubMed.
- N. Monteiro, M. Martins, A. Martins, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, Acta Biomater., 2015, 18, 196–205 CrossRef CAS PubMed.
- N. Monteiro, A. Martins, R. Pires, S. Faria, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, Biomater. Sci., 2014, 2, 1195–1209 RSC.
- N. Liu and H.-J. Park, Colloids Surf., B, 2010, 76, 16–19 CrossRef CAS PubMed.
- J. N. Moreira, T. Ishida, R. Gaspar and T. M. Allen, Pharm. Res., 2002, 19, 265–269 CrossRef CAS.
- L. C. Gomes-da-Silva, A. O. Santos, L. M. Bimbo, V. Moura, J. S. Ramalho, M. C. P. de Lima, S. Simoes and J. N. Moreira, Int. J. Pharm., 2012, 434, 9–19 CrossRef CAS PubMed.
- K. Katayama, Y. Kato, H. Onishi, T. Nagai and Y. Machida, Int. J. Pharm., 2002, 248, 93–99 CrossRef CAS PubMed.
- K. Yamabe, Y. Kato, H. Onishi and Y. Machida, J. Controlled Release, 2003, 90, 71–79 CrossRef CAS PubMed.
- Q. Wang, E. C. Rojas and K. D. Papadopoulos, J. Colloid Interface Sci., 2012, 383, 89–95 CrossRef CAS PubMed.
- N. K. Satija, G. U. Gurudutta, S. Sharma, F. Afrin, P. Gupta, Y. K. Verma, V. K. Singh and R. P. Tripathi, Stem Cells Dev., 2007, 16, 7–23 CrossRef CAS PubMed.
- C. Vater, P. Kasten and M. Stiehler, Acta Biomater., 2011, 7, 463–477 CrossRef CAS PubMed.
- N. Monteiro, E. E. Smith, S. Angstadt, W. Zhang, A. Khademhosseini and P. C. Yelick, Biomaterials, 2016, 106, 167–179 CrossRef CAS PubMed.
- S. Liao, C. K. Chan and S. Ramakrishna, Mater. Sci. Eng., C, 2008, 28, 1189–1202 CrossRef CAS.
- D. M. Huang, Y. Hung, B. S. Ko, S. C. Hsu, W. H. Chen, C. L. Chien, C. P. Tsai, C. T. Kuo, J. C. Kang, C. S. Yang, C. Y. Mou and Y. C. Chen, FASEB J., 2005, 19, 2014–2020 CrossRef CAS PubMed.
- A. Martins, A. R. C. Duarte, S. Faria, A. P. Marques, R. L. Reis and N. M. Neves, Biomaterials, 2010, 31, 5875–5885 CrossRef CAS PubMed.
- A. O. El-Sadik, A. El-Ansary and S. M. Sabry, Clin. Pharmacol.: Adv. Appl., 2010, 2, 9–16 CAS.
- N. Monteiro and P. Yelick, J. Tissue Eng. Regener. Med., 2016 DOI:10.1002/term.2134..
- I. Pountos, D. Corscadden, P. Emery and P. V. Giannoudis, Injury, 2007, 38, S23–S33 CrossRef PubMed.
- T. H. Park, H. J. Lee, J. A. Kim and S. H. Lee, J. Biotechnol., 2007, 131, S65 CrossRef.
- N. Jaiswal, S. E. Haynesworth, A. I. Caplan and S. P. Bruder, J. Cell. Biochem., 1997, 64, 295–312 CrossRef CAS PubMed.
- V. E. Santo, K. Sato, J. Ratanavaraporn, M. E. Gomes, J. F. Mano, R. L. Reis and Y. Tabata, J. Tissue Eng. Regener. Med., 2012, 6, 330 Search PubMed.
- J. M. Oliveira, N. Kotobuki, M. Tadokoro, M. Hirose, J. F. Mano, R. L. Reis and H. Ohgushi, Bone, 2010, 46, 1424–1435 CrossRef CAS PubMed.
- G. Z. Jin, M. Eltohamy and H. W. Kim, RSC Adv., 2015, 5, 26832–26842 RSC.
- P. Kallinteri, S. G. Antimisiaris, D. Karnabatidis, C. Kalogeropoulou, I. Tsota and D. Siablis, Biomaterials, 2002, 23, 4819–4826 CrossRef CAS PubMed.
- S. Abbas, C. Da Wei, K. Hayat and X. Zhang, Food Rev. Int., 2012, 28, 343–374 CrossRef CAS.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- A. Murao, M. Nishikawa, C. Managit, J. Wong, S. Kawakami, F. Yamashita and M. Hashida, Pharm. Res., 2002, 19, 1808–1814 CrossRef CAS.
- V. P. Torchilin and V. Weissig, Liposomes: A Practical Approach, Oxford University Press, 2nd edn, 2003 Search PubMed.
- N. A. Peppas and L. Brannonpeppas, J. Food Eng., 1994, 22, 189–210 CrossRef.
- B. Delorme and P. Charbord, Methods Mol. Med., 2007, 140, 67–81 CAS.
- K.-M. Choi, Y.-K. Seo, H.-H. Yoon, K.-Y. Song, S.-Y. Kwon, H.-S. Lee and J.-K. Park, J. Biosci. Bioeng., 2008, 105, 586–594 CrossRef CAS PubMed.
- S. A. Asgeirdottir, J. Kamps, H. I. Bakker, P. J. Zwiers, P. Heeringa, K. van der Weide, H. van Goor, A. H. Petersen, H. Morselt, H. E. Moorlag, E. Steenbergen, C. G. Kallenberg and G. Molema, Mol. Pharmacol., 2007, 72, 121–131 CrossRef PubMed.
- N. Monteiro, A. Martins, D. Ribeiro, S. Faria, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, J. Tissue Eng. Regener. Med., 2013, 9, 1056–1066 CrossRef PubMed.
- E. Gibbons, M. C. Allwood, T. Neal and G. Hardy, J. Pharm. Biomed. Anal., 2001, 25, 605–611 CrossRef CAS PubMed.
- L. Wechtersbach, N. P. Ulrih and B. Cigic, LWT--Food Sci. Technol., 2012, 45, 43–49 CrossRef CAS.
- W. M. Nau, J. Am. Chem. Soc., 1998, 120, 12614–12618 CrossRef CAS.
- P. J. Jansson, H. R. Jung, C. Lindqvist and T. Nordstrom, Free Radical Res., 2004, 38, 855–860 CrossRef CAS PubMed.
- N. I. DeNardis, I. Ruzic, J. Pecar-Ilic, S. El Shawish and P. Ziherl, Bioelectrochemistry, 2012, 88, 48–56 CrossRef PubMed.
- V. A. Tsotas, S. Mourtas and S. G. Antimisiaris, Drug Delivery, 2007, 14, 441–445 CrossRef CAS PubMed.
- D. C. Drummond, O. Meyer, K. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–744 CAS.
- F. Szoka Jr and D. Papahadjopoulos, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4194–4198 CrossRef CAS.
- N. J. Zuidam, H. K. M. E. Gouw, Y. Barenholz and D. J. A. Crommelin, Biochim. Biophys. Acta, Biomembr., 1995, 1240, 101–110 CrossRef.
- L. M. Ickenstein, M. C. Sandström, L. D. Mayer and K. Edwards, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 171–180 CrossRef CAS PubMed.
- A. Yousefi, F. Esmaeili, S. Rahimian, F. Atyabi and R. Dinarvand, Sci. Pharm., 2008, 453–464, DOI:10.3797/scipharm.0806–08.
- M. Barzegar-Jalali, K. Adibkia, H. Valizadeh, M. R. S. Shadbad, A. Nokhodchi, Y. Omidi, G. Mohammadi, S. H. Nezhadi and M. Hasan, J. Pharm. Pharm. Sci., 2008, 11, 167–177 CAS.
- M. M. Nounou, L. K. El-Khordagui, N. A. Khalafallah and S. A. Khalil, Acta Pharm., 2006, 56, 311–324 CAS.
- S. Kawakami, A. Sato, M. Yamada, F. Yamashita and M. Hashida, STP Pharma Sci., 2001, 11, 117–120 CAS.
- V. E. Santo, M. E. Gomes, J. F. Mano and R. L. Reis, Tissue Eng., Part B, 2012, 19(4), 308–326 CrossRef PubMed.
- G. R. Kirkham and S. H. Cartmell, in Topic in Tissue Engineering, 2007, vol. 3, ch. 13 Search PubMed.
- Y. Zhang, X. Deng, E. L. Scheller, T.-G. Kwon, J. Lahann, R. T. Franceschi and P. H. Krebsbach, Biomaterials, 2010, 31, 3231–3236 CrossRef CAS PubMed.
- R. M. Porter, W. R. Huckle and A. S. Goldstein, J. Cell. Biochem., 2003, 90, 13–22 CrossRef CAS PubMed.
- B. C. Dash, G. Rethore, M. Monaghan, K. Fitzgerald, W. Gallagher and A. Pandit, Biomaterials, 2010, 31, 8188–8197 CrossRef CAS PubMed.
- J. Senior, J. C. W. Crawley and G. Gregoriadis, Biochim. Biophys. Acta, 1985, 839, 1–8 CrossRef CAS.
- K. Kettler, P. Krystek, C. Giannakou, A. J. Hendriks and W. H. de Jong, J. Nanopart. Res., 2016, 18, 182 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |