
Identification of a novel selective inhibitor of mutant isocitrate dehydrogenase 1 at allosteric site by docking-based virtual screening†
Fangxia Zou‡
ab,
Stefan Pusch‡cd,
Jessica Eiselc,
Tianfang Maab,
Qihua Zhue,
Dawei Dengb,
Yueqing Gub,
Yungen Xue,
Andreas von Deimling*cd and
Xiaoming Zha*ab
aJiangsu Key Laboratory of Drug Screening, State Key Laboratory of Natural Medicines, China. E-mail: xmzha@cpu.edu.cn; Fax: +86-25-83271142; Tel: +86-25-83271057
bDepartment of Biochemical Engineering, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, PR China
cGerman Consortium of Translational Cancer Research (DKTK), Clinical Cooperation Unit Neuropathology, German Cancer Research Center (DKFZ), INF 280, Heidelberg D-69120, Germany. E-mail: andreas.vondeimling@med.uni-heidelberg.de; Fax: +49-6221-56-4655; Tel: +49-6221-56-4650
dDepartment of Neuropathology, Institute of Pathology, Ruprecht-Karls-University Heidelberg, INF 224, Heidelberg D-69120, Germany
eDepartment of Medicinal Chemistry, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, PR China
First published on 26th September 2016
Abstract
Isocitrate dehydrogenase 1 (IDH1), catalyzing oxidative decarboxylation of isocitrate to provide energy for aerobic organisms, is an essential enzyme in the tricarboxylic acid cycle. However, mutant IDH1 (mIDH1) produces oncometabolite D-2-hydroxyglutarate (D2HG) and has recently been confirmed in several types of cancers, particularly in glioma and acute myeloid leukemia. Herein a docking-based virtual screening (VS) of SPECS library was conducted for the allosteric site of mIDH1. The cellular evaluation of the hit compounds led to the identification of FX-03 as a novel selective mIDH1 inhibitor at allosteric site with IC50 values of 55.50 μM and 68.38 μM in HEK-293T cells transfected with IDH1 R132H and IDH1 R132C, respectively. Importantly, FX-03 owned significant selectivity with no inhibition in HEK-293T cells transfected with IDH1 WT. These findings indicate that VS of mIDH1's allosteric site represents a useful strategy for discovery of selective mIDH1 inhibitors and FX-03 deserves further optimization as a lead compound in future study.
Introduction
Utilizing NADP+ and Mg2+ as cofactors, isocitrate dehydrogenase 1 (IDH1) catalyzes oxidative decarboxylation of isocitrate acid to α-ketoglutaric acid (α-KG), one of the key reactions in the tricarboxylic acid (TCA) cycle, which providing majority of energy in aerobic organisms derived from fats or carbohydrates, as shown in Fig. 1.1 This enzymatic pathway regulates cellular epigenetic state related to various molecular processes.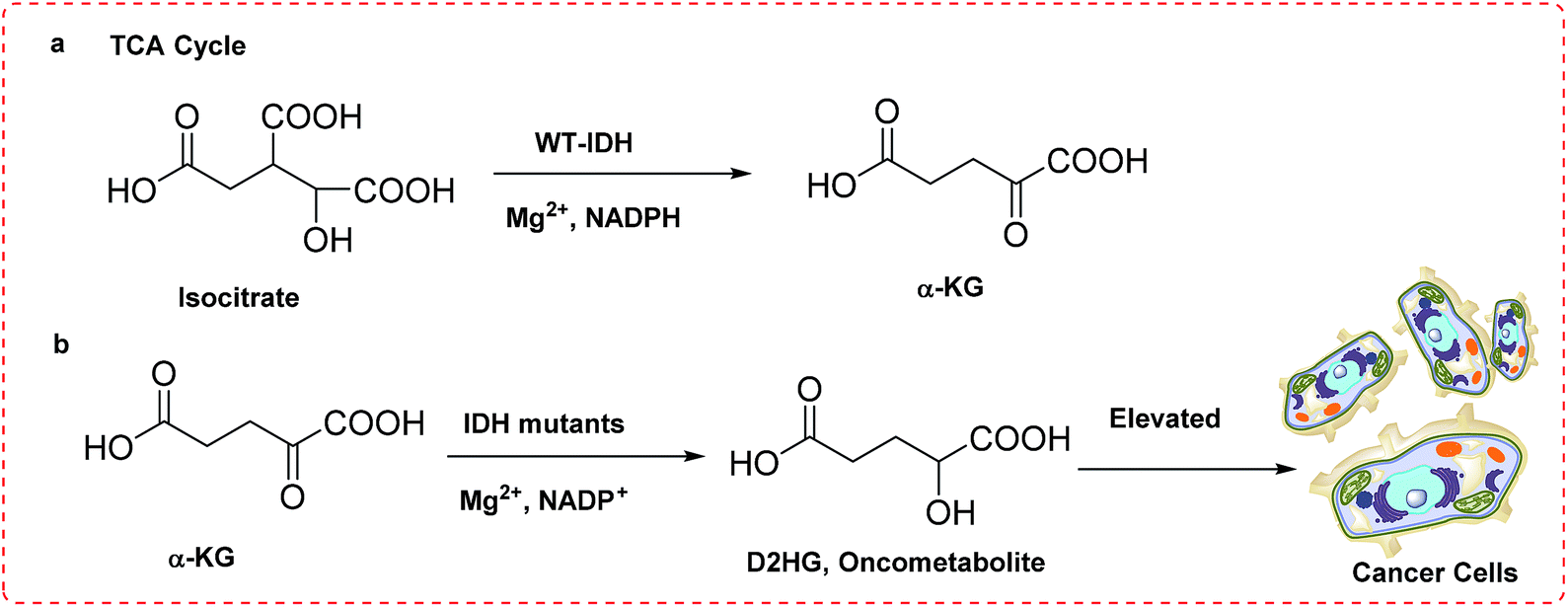 | ||
| Fig. 1 (a) Reaction catalyzed by IDH1 WT in TCA cycle; (b) reaction catalyzed by mIDH1 initiating tumorgenesis. | ||
Three IDH isoforms do exist in humans, with IDH1 locates in the cytosol and peroxisome, IDH2 and IDH3 in mitochondria.2,3 However, strong evidences showed that somatic mutations of IDH1 conferred a neomorphic enzymatic activity associated with several types of tumors. For example, mutation in IDH1 was founded in ∼75% low-grade II and III gliomas, as well as secondary glioblastoma multiforme developed from low-grade tumors with 5 year survival of <10%.4,5 A special feature has been particularly noted that mutation of IDH1 is exclusively located in a key arginine residue belonging to the active site of enzyme, with the sign of R132H being the predominant (>90%).6,7
Structural alignments of IDH1 R132H and IDH1 WT (Fig. 2) indicate that IDH1 R132H has two ligand binding sites, site I and site II, with the binding site II being the catalytic site ∼6.5 Å from the site I, however, IDH1 WT only has the binding site II. Isocitrate is located in site II of IDH1 WT and binds to site I of IDH1 R132H, while α-KG occupies the catalytically active binding site II. It is noted that the two binding sites, site I and II in IDH1 R132H do not coexist, explaining why IDH1 R132H has a gain of function activity in reduction of α-KG to the oncometabolite D-2-hydroxyglutaric acid (D2HG). In addition, normal cells have low levels of D2HG, whereas cells harboring IDH1 mutation exhibits the significantly elevated concentrations of D2HG. High concentrations of D2HG have been characterized in tumors with IDH1 point mutation.8–10 For example, 100-fold elevated levels of D2HG have been detected in the glioma patient samples containing mutant IDH1.11
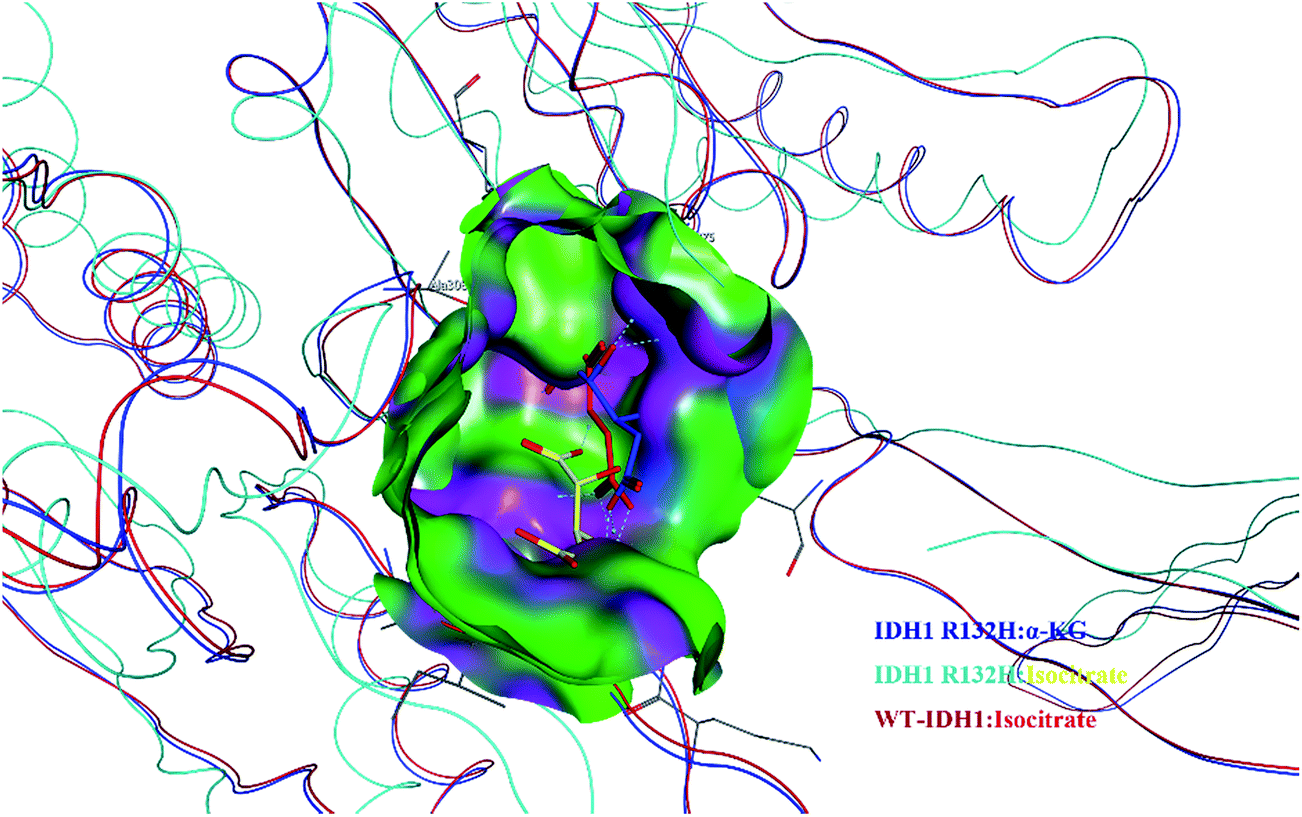 | ||
| Fig. 2 Close-up, view the positions of the aligned structures of IDH1 R132H: α-KG (in blue), IDH1 R132H: isocitrate (in yellow), and IDH1 WT: isocitrate (in red). | ||
Furthermore, it is demonstrated that D2HG is a competitive inhibitor of many α-KG dependent dioxygenases, including DNA and histone demethylases. This inhibition leads to the tumorgenesis contributing to the block of cell differentiation and the hypermethylation of DNA/histone through epigenetic rewiring.12 Therefore, high levels of D2HG from IDH1 somatic point mutations are responsible for the initiation and progression of cancers.13,14 Taken these genetic findings together, it is implicated that oncogenic mIDH1 is a compelling drug target for intervention in cancer treatment.
Owing to the potential of mIDH1 inhibitors as anti-tumor therapeutics, a variety of researchers have investigated the structural and mechanistic aspects of mIDH1 inhibition. Due to the responsibility of wild-type IDH1 enzyme in primary metabolism, the critical issue in designing mIDH1 inhibitor is to achieve selective inhibition of the mutant enzyme over the wild-type. Several chemo-types of mIDH1 inhibitors displayed in Fig. 3 have been reported to date showing highly on-target activity towards reducing the cellular production of D2HG.15 AGI-5198 (1), also known as IDH-C35, is the first potent and selective allosteric mIDH1 inhibitor of IDH1 R132H and R132C with IC50 values of 70 nM and 160 nM. It was shown to have potential non-competitive inhibition versus α-KG substrate.16 1-Hydroxypyridin-2-one core of compounds (2 and 3), binding to an orthosteric isocitrate active site, were identified as competitive and potent inhibitors of IDH1 R132H with their Ki values of 190 nM and 280 nM, respectively, and have no effects on homodimer IDH1 WT.17 However, no similar ligand-bound crystal complex was reported, and the mechanism of inhibition was investigated to be uncompetitive with NADPH. The derivatives holding phenyl-glycine scaffold such as 4 and 5 showed ∼90% tumor D2HG inhibition in vivo mouse xenograft following repeat dosing.18 GSK321 (6) was found to be a highly potent mIDH1 inhibitor with the IC50 values of 4.6 nM, 3.8 nM, and 2.9 nM against IDH1 R132H, IDH1 R132C and IDH1 R132G, respectively, as well as minimal inhibitory activity against mutant IDH2.19 6 does not bind at the same pocket as 2 and 3 do, and displays a non-competitive mode of inhibition versus NADPH and a competitive inhibition relationship with α-KG substrate. The first co-crystal structure of mIDH1 inhibitor complex (VVS in PDB code 4UMX) was published by Deng and his colleagues.20 VVS (7) contained an imidazole group protruding into each subunit of IDH1 R132H homodimer to make direct interactions with the residue of Asp279 in the metal-binding pocket. 7 was a time-dependent and reversible mIDH1 inhibitor with IC50 value of 13 nM for IDH1 R132H.
 | ||
| Fig. 3 The structures and inhibitory activities of reported mIDH1 inhibitors. | ||
Recent studies disclosed that Mg2+ was much more effective on saturating its allosteric site (also called as divalent magnesium binding site of mIDH1) in the wild-type IDH1 than the mutant one. Therefore, competitive binding with Mg2+ at the allosteric site may contribute to the selectivity of mIDH1 inhibitors. In this paper, focusing on the allosteric site of mIDH1, we reported a docking-based VS with three docking softwares including HTVS, SP and XP in Glide v5.5,21–23 combined with the subsequent biological experiments to identify the mIDH1 inhibitors with novel skeletons. The process was described in Fig. 4. Finally, four compounds were identified to have with micromolar activities towards reduction of oncometabolite D2HG and among which, FX-03 was further confirmed as a selective mIDH1 inhibitor for further optimization. These results preliminarily pave the way for discovery and development of novel selective mIDH1 inhibitors at allosteric site for cancer treatment.
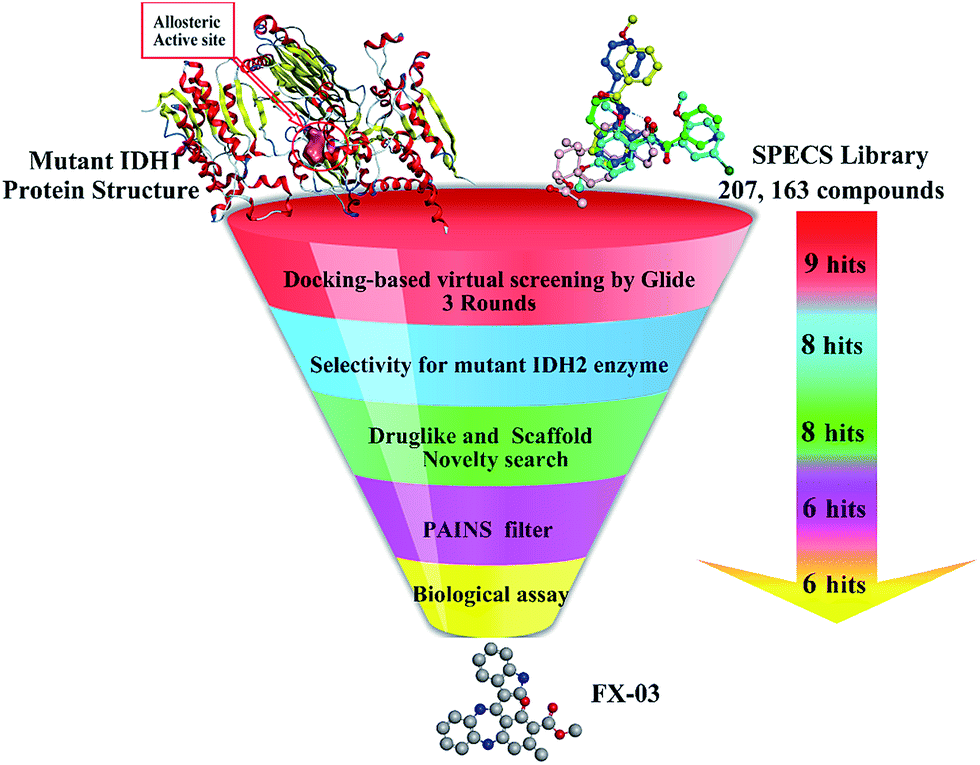 | ||
| Fig. 4 The flowchart combined with docking-based VS and bioassays for mIDH1 inhibitors. | ||
Results and discussion
Virtual screening (VS)
In an effort to identify available chemical and structural information of promising hits for IDH1 R132H, a three-round of docking based VS strategy was employed to filter out molecules lacking essential binding features at allosteric site. The ligand database was extracted from the SPECS library with over 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules. The initial potential energy of the ligand was minimized by Pipeline Pilot 8.0, followed with submission to Ligprep module to generate the indexed stereoisomers with OPLS_2005 force field and possible ionization state with Epik module (Maestro, Schrödinger v2015). Thus 207163 molecules were well prepared and subjected to three rounds of docking-based VS. The model of VVS (7) binding with recombinant IDH1 R132H homodimer (PDB code 4UMX) was selected for subsequent docking procedure.
000 molecules. The initial potential energy of the ligand was minimized by Pipeline Pilot 8.0, followed with submission to Ligprep module to generate the indexed stereoisomers with OPLS_2005 force field and possible ionization state with Epik module (Maestro, Schrödinger v2015). Thus 207163 molecules were well prepared and subjected to three rounds of docking-based VS. The model of VVS (7) binding with recombinant IDH1 R132H homodimer (PDB code 4UMX) was selected for subsequent docking procedure.
To understand the interaction mode of mIDH1 binding cavity, the VVS–mIDH1 complex was thoroughly checked and analyzed. VVS bound to one subunit near the dimer interface, consistent with an allosteric mode of inhibition. The metal-binding Asp279 residue making a direct interaction with VVS, which disrupting catalytically required elements in the other subunit.
To select an appropriate docking tool, three docking programs (GOLD, CDOCKER and Glide) were employed by evaluating their reproducing ability in VVS redocking procedure. Eventually Glide v5.5 was chosen as a predictable tool due to its most similar bioactive binding character with the RMSD values of 0.32 Å (Glide HTVS mode), 0.25 Å (Glide SP mode) and 0.13 Å (Glide XP mode), respectively. Then HTVS, SP and XP procedures integrated in Glide protocol were successively utilized to dock the prepared SPECS library into mIDH1's allosteric site. Asp279 in one subunit was reported to be involved in the specific recognition of VVS in dimer surface, therefore, it was regarded as a key polar hydrogen bond constraint during the whole Glide docking procedure. Apart from the bond interaction in binding site criterion, both XP GScore −9 kJ mol−1 and Glide Emodel energy −72 kJ mol−1 were set as the cutoff values, and the XP GScore and Glide Emodel energy values of VVS redocking results were −8.698 kJ mol−1 and −71.653 kJ mol−1, respectively. Docking studies were performed with Glide software with Glide HTVS mode at first, the top 10% compounds (9761 hits) from HTVS were redocked with the more computational expensive Glide SP scoring module. This led to selection of 10% top-ranked compounds (837 hits) filtering for Glide extra precision (XP) and scoring methods. Eventually, nine top-ranked molecules showing favorable interaction with mIDH1 in proper orientation and with good docking scores were retrieved and were furtherly submitted in selectivity checking for mutant IDH2 enzyme.
Cellular investigation showed that Km value of mIDH1 catalytic reaction would be markedly higher (>300-fold) than the one of wild-type IDH1 catalytic reaction, indicating that Mg2+ was more effectively rephrasing towards mIDH1's allosteric site. Under such conditions, competitive inhibition at allosteric site existed in wild type homodimer would be too negligible. In terms of selectivity of mIDH1 over mutant IDH2 (mIDH2), docking was implemented with the co-crystal structure of IDH2 R140Q and AGI-6780 (PDB code 4JA8) utilizing Glide XP (Schrödinger v2015) according to the re-docking performances. Just consider the binding features, eight compounds performed poorly, with the molecular orientations and extensive hydrogen bond interactions in this distinct allosteric site on IDH2 which were much worse than the initial ligand AGI-6780. Therefore, eight hits were furtherly considered for drug-likeness studies.
Drug-likeness properties are an important indicator for selecting the compounds for further in vitro studies, which includes molecular or physicochemical properties that contribute to favorable “Lipinski's rule of five” and ADMET filter in DS3.0. ADMET descriptor values were set as AlogP98 ≥ 3, logS ≥ −6 and PSA ≤ 112 for prerequisite. Based on drug-likeness check, finally eight compounds were all kept and submitted to similarity evaluation with several different series of the reported mIDH1 inhibitors (Table S1†). Visualizing the results of the compare libraries protocol in Pipeline Pilot 8.0, the number of global fingerprint bits only in Library A was 8, number of global fingerprint bits only in Library B was 7, the similarity score and Bayesian distance between the two libraries was 0.4667 and 90.7856, respectively. These results correlated the scaffold novelty of eight compounds. After removal of the PAINS compounds online, six hits (schemed in Fig. 5) were selected and purchased for further biological assays.
 | ||
| Fig. 5 The structures of six retrieved compounds. | ||
Cellular assays
| Compd | Glide GScore_energy (kJ mol−1) | Emodel energy (kJ mol−1) | D2HG reduction (%, IDH1 R132H) | D2HG reduction (%, IDH1 R132C) | ||
|---|---|---|---|---|---|---|
| 5 μM | 50 μM | 5 μM | 50 μM | |||
| FX-01 | −10.015 | −87.455 | 6.2 | 34.7 | 1.2 | 22.3 |
| FX-02 | −9.920 | −75.271 | 17.0 | 54.5 | 14.2 | 45.2 |
| FX-03 | −9.353 | −73.682 | 18.1 | 43.5 | 5.7 | 40.0 |
| FX-04 | −9.464 | −72.670 | −1.3 | 28.3 | −7.4 | 20.5 |
| FX-05 | −10.178 | −83.905 | −1.2 | 43.1 | −10.3 | 27.6 |
| FX-06 | −9.037 | −80.147 | 20.4 | 97.4 | 6.8 | 95.7 |
| VVS | −8.698 | −8.698 | ND | ND | ND | ND |
| AGI-5198 | ND | ND | 98.7 | 99.6 | 86.7 | 99.1 |
Cytotoxicity towards HEK-293T IDH1 WT
It is of importance that an ideal mIDH1 inhibitor should own weak or no effect toward wild-type IDH1. To investigate the selectivity of mIDH1 over IDH1 WT, we thereby used four compounds (FX-01, FX-02, FX-03 and FX-06) that have shown their inhibitory potential to test their viability of HEK-293T cells expressing IDH1 WT. In contrast to FX-01, FX-02 and FX-06, FX-03 showed no significant toxicity at all tested concentrations (Fig. 6). | ||
| Fig. 6 Cytotoxicity of FX-01, FX-02, FX-03 and FX-06 in different concentrations. Indicated numbers depict the % toxicity at the concentrations used in the D2HG assay. | ||
Determination of cellular IC50 of FX-03
With FX-03 in hand, we further determined its cellular IC50 values of HEK-293T cells transfected with IDH1 R132H and IDH1 R132C, which were 55.50 μM and 68.38 μM, respectively (Fig. 7). | ||
| Fig. 7 Potential inhibition of mIDH1 enzyme leads to decreased production of intracellular 2-HG in HEK-293T cells transfected with IDH1 R132H and IDH1 R132C after treatment with FX-03 for 3 days. Half-maximal inhibitory concentration values for decreased intracellular 2-HG levels are indicated. Graphs are representative of three independent experiments. | ||
Binding feature of FX-03
To understand the mechanism basis of the inhibitory activity towards mIDH1, we analyzed the best putative binding mode of FX-03 via eye checking and critical residues modeling. In the binding mode of FX-03 with IDH1 R132H homodimer, hydrogen bond interaction was observed between the nitrogen atom of 1,4-diazepine core and the carbonyl group of Asp279. It favored the hydrogen bond between the initial crystal ligand VVS and the allosteric site of IDH1 R132H (Fig. 8A). Particularly, in the binding mode of FX-03, a hydrogen bond was formed between the amine group of indole ring and Val255 in one subunit and a π–π stacking interaction between the indole group and Trp267 (Fig. 8B). The lengths of two hydrogen bonds were 2.3 Å and 2.0 Å, respectively, indicating that hydrogen bonds were essential and strong for the inhibitory activities of IDH1 R132H, IDH1 R132C and IDH1 R100Q. Subsequently, docking protocol was employed to visualize the favorable bond interactions between FX-03 and the active site of IDH2 R140Q. As supplemented in Fig. 8C and D, the hydrogen bond between FX-03 and IDH2 R140Q was not formed with key residue Gln316 of two subunits, while Gln316 was an important residue for the binding mode of mIDH2 inhibitors.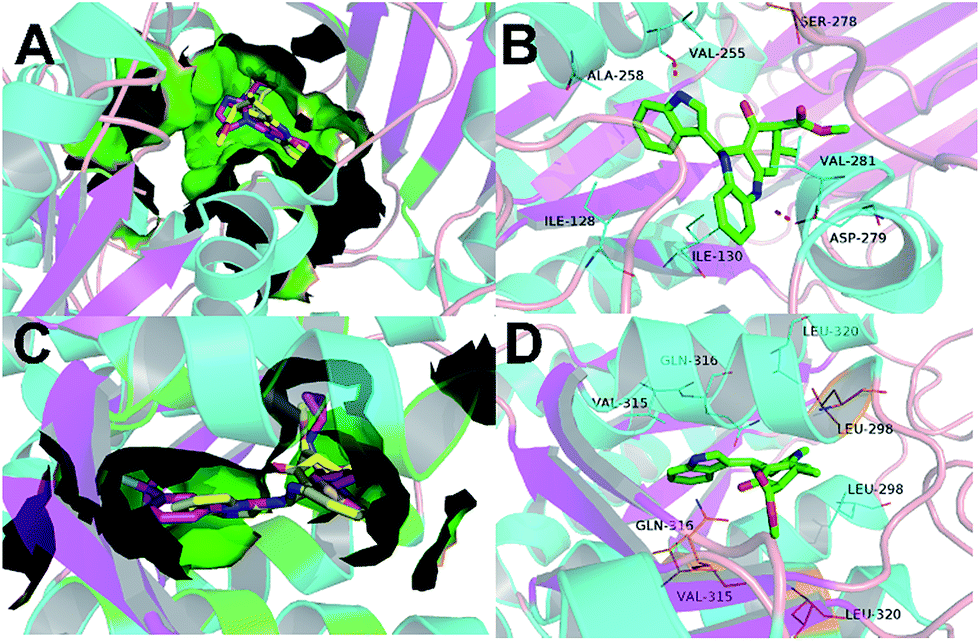 | ||
| Fig. 8 (A) The superimposed structures of homodimer IDH1 R132H in complex with VVS (with C atoms in purple) and FX-03 (in yellow). (B) Binding mode of FX-03 (PDB code 4UMX). FX-03 is displayed in green, oxygen atoms are in red and nitrogen atoms in blue. Hydrogen bonds between homodimer mutant IDH1 and FX-03 are represented as a red dash line. All of the compounds and residues are labeled. (C) Superimposed structures of homodimer IDH2 R140Q in complex with AGI-6780 (with C atoms in purple) and FX-03 (in yellow). (D) Binding mode of FX-03 (PDB code 4UMX). FX-03 is displayed in green, oxygen atoms are in red and nitrogen atoms in blue. Hydrogen bonds between homodimer mIDH2 and FX-03 are represented as a red dash line. All of the compounds and residues are labeled. | ||
Conclusions
Biochemical studies have verified that mIDH1 owned a new function in reduction of α-KG to D2HG, an elevated oncometabolite accumulation in cancer patients for inhibition of the normal histone lysine demethylation and a hindrance of cell differentiation that eventually cause tumor initiation. Further pharmacological investigation demonstrated that mIDH1 was a compelling drug target for several types of cancer. To date, only limited scaffold chemotypes of mIDH1 inhibitors have been reported and mostly are competitive with isocitrate or α-KG, resulted in their poor structural novelty and low specificity. Upon docking-based VS of mIDH1' allosteric site, selectivity checking on related homologous mIDH2, drug-like properties estimating, PAINS filtering and novelty searching procedures, six compounds were retrieved to test their biological activities. Four compounds FX-01, FX-02, FX-03 and FX-06 displayed remarkable inhibitions in 50 μM concentrations against the production of D2HG, which was excreted from HEK-293T cells transfected with IDH1 R132H and IDH1 R132C. Importantly, only FX-03 further displayed nearly no cytotoxicity toward HEK-293T cells expressing IDH1 WT, however, showing its IC50 values of 55.50 μM and 65.38 μM in HEK-293T cells transfected with IDH1 R132H and IDH1 R132C, respectively. These findings indicate that VS of mIDH1's allosteric site represents a useful strategy for discovery of mIDH1 inhibitor and FX-03 deserves further optimization as a selective mIDH1 inhibitor at the allosteric site in future study.Experimental section
Computational methods
000 ligands extracted from the SPECS library were submitted to ligand preparation protocol of Maestro 10.0 to generate the stereoisomers and possible ionization state with Epik, following with final optimized by OPLS_2005 force field.24,25 Eventually, a total database of 207163 molecules was prepared well as the starting point for docking-based VS.To validate the optimal chosen docking protocol, RMSD values were utilized to evaluate the performance differences of several docking modules, containing CDOCKER/DS 3.0, GOLD v5.0 and Glide implemented in Schrödinger v2015. Docking procedures were verified by reproducing initial crystal ligand VVS to IDH1 R132H homodimer. Finally, Grid module was characterized for the generation of grid file and the receptor grid was defined by an enclosing box that centered on VVS. The module automatically optimize hydroxyl, Asn, Gln, and His states using ProAssign, and restrained minimization using OPLS_2005 force field. Flips of 5- and 6-member rings were allowed and non-polar of amide bonds was penalized. Writ out at most 1 pose per ligand. Docking studies were performed with Glide software with Glide High Throughout Virtual Screening (HTVS) at first, proven to discard noticeable non-binders with minimal computational time, and then the top 10% compounds (∼10000) from HTVS were redocked with the more computational expensive Glide standard precision (SP) scoring module. This led to selection of 10% top-ranked compounds filtering for Glide extra precision (XP) and scoring methods. Top-ranked compounds with XP_GScore or Glide Emodel low than VVS redocking scoring values were clustered for visual inspection of the conformation features and important relationships with surrounding residues.
In order to define the high selectivity of these ligands towards mIDH2, accurate structure of IDH2 R140Q complex (PDB code 4JA8) was set for grid calculation. The screening against IDH2 R140Q at the desired grid coordinates was performed through exact precision (XP) docking algorithm. Nine ligands were treated as flexible, followed by flips of 5- and 6-member rings and penalization of conformation of non-planar amide bonds. Mostly, one pose for each ligand was exported and the remained parameters were referred to keep default set.26,27
P ≤ 5. The four above physicochemical parameters are closely associated with acceptable intestinal permeability and aqueous solubility, which can help us reduce interference in the design and development of lead compounds. Besides, the ADMET protocol in DS3.0 was utilized to calculate their physiochemical descriptors and required principles of possible hits with detailed oral absorbability such as AlogP98, logS and PSA.Biological assay
000 cells per well. Subsequently they were treated with compound at the depicted concentration and incubated for 72 h. There after the cell viability was analyzed using CellTiterGlo (Promega, Madison, USA) following the manufacturers protocol.Conflict of interest
Prof. Andreas von Deimling and Dr Stefan Pusch are patent holders for the enzymatic 2-HG detection assay. All other authors declare no competing financial interest.Abbreviations
| IDH | Isocitrate dehydrogenase |
| ICT | Isocitrate acid |
| α-KG | α-Ketoglutaric acid |
| D2HG | D-2-Hydroxyglutaric acid |
| mIDH1 | Mutant IDH1 |
| mIDH2 | Mutant IDH2 |
| R132H | Arg132 mutation to His |
| R132C | Arg132 mutation to Cys |
| WT | Wild type |
| R140Q | Arg140 mutation to Gln |
Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province of China (BK20161458), the Open Project Program of Jiangsu Key Laboratory of Drug Screening (JKLDS2015KF-03) and the Graduate Innovative Foundation supported by Huahai Pharmaceuticals Co., Ltd. (CX14S-004HH).Notes and references
- X. Xu, J. Zhao, Z. Xu, B. Peng, Q. Huang, E. Arnold and J. Ding, J. Biol. Chem., 2004, 279, 33946–33957 CrossRef CAS PubMed.
- Z. Liu, Y. Yao, M. Kogiso, B. Zheng, L. Deng, J. J. Qiu, S. Dong, H. Lv, J. M. Gallo, X. N. Li and Y. Song, J. Med. Chem., 2014, 57, 8307–8318 CrossRef CAS PubMed.
- L. Li, A. C. Paz, B. A. Wilky, B. Johnson, K. Galoian, A. Rosenberg, G. Hu, G. Tinoco, O. Bodamer and J. C. Trent, PLoS One, 2015, 10, e0133813 Search PubMed.
- D. W. Parsons, S. Jones, X. Zhang, J. C. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, I. M. Siu, G. L. Gallia, A. Olivi, R. McLendon, B. A. Rasheed, S. Keir, T. Nikolskaya, Y. Nikolsky, D. A. Busam, H. Tekleab, L. A. Diaz Jr, J. Hartigan, D. R. Smith, R. L. Strausberg, S. K. Marie, S. M. Shinjo, H. Yan, G. J. Riggins, D. D. Bigner, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V. E. Velculescu and K. W. Kinzler, Science, 2008, 321, 1807–1812 CrossRef CAS PubMed.
- M. F. Amary, K. Bacsi, F. Maggiani, S. Damato, D. Halai, F. Berisha, R. Pollock, P. O'Donnell, A. Grigoriadis, T. Diss, M. Eskandarpour, N. Presneau, P. C. Hogendoorn, A. Futreal, R. Tirabosco and A. M. Flanagan, J. Pathol., 2011, 224, 334–343 CrossRef CAS PubMed.
- W. Xu, H. Yang, Y. Liu, Y. Yang, P. Wang, S. H. Kim, S. Ito, C. Yang, P. Wang, M. T. Xiao, L. X. Liu, W. Q. Jiang, J. Liu, J. Y. Zhang, B. Wang, S. Frye, Y. Zhang, Y. H. Xu, Q. Y. Lei, K. L. Guan, S. M. Zhao and Y. Xiong, Cancer Cell, 2011, 19, 17–30 CrossRef CAS PubMed.
- A. R. Rendina, B. Pietrak, A. Smallwood, H. Zhao, H. Qi, C. Quinn, N. D. Adams, N. Concha, C. Duraiswami, S. H. Thrall, S. Sweitzer and B. Schwartz, Biochemistry, 2013, 52, 4563–4577 CrossRef CAS PubMed.
- S. Gross, R. A. Cairns, M. D. Minden, E. M. Driggers, M. A. Bittinger, H. G. Jang, M. Sasaki, S. Jin, D. P. Schenkein, S. M. Su, L. Dang, V. R. Fantin and T. W. Mak, J. Exp. Med., 2010, 207, 339–344 CrossRef CAS PubMed.
- T. C. Pansuriya, R. van Eijk, P. d'Adamo, M. A. van Ruler, M. L. Kuijjer, J. Oosting, A. M. Cleton-Jansen, J. G. van Oosterwijk, S. L. Verbeke, D. Meijer, T. van Wezel, K. H. Nord, L. Sangiorgi, B. Toker, B. Liegl-Atzwanger, M. San-Julian, R. Sciot, N. Limaye, L. G. Kindblom, S. Daugaard, C. Godfraind, L. M. Boon, M. Vikkula, K. C. Kurek, K. Szuhai, P. J. French and J. V. Bovee, Nat. Genet., 2011, 43, 1256–1261 CrossRef CAS PubMed.
- B. Pietrak, H. Zhao, H. Qi, C. Quinn, E. Gao, J. G. Boyer, N. Concha, K. Brown, C. Duraiswami, R. Wooster, S. Sweitzer and B. Schwartz, Biochemistry, 2011, 50, 4804–4812 CrossRef CAS PubMed.
- L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin, M. C. Keenan, K. M. Marks, R. M. Prins, P. S. Ward, K. E. Yen, L. M. Liau, J. D. Rabinowitz, L. C. Cantley, C. B. Thompson, M. G. Vander Heiden and S. M. Su, Nature, 2010, 465, 966 CrossRef CAS PubMed.
- C. Lu, P. S. Ward, G. S. Kapoor, D. Rohle, S. Turcan, O. Abdel-Wahab, C. R. Edwards, R. Khanin, M. E. Figueroa, A. Melnick, K. E. Wellen, D. M. O'Rourke, S. L. Berger, T. A. Chan, R. L. Levine, I. K. Mellinghoff and C. B. Thompson, Nature, 2012, 483, 474–478 CrossRef CAS PubMed.
- K. Garber, J. Natl. Cancer Inst., 2010, 102, 926–928 CrossRef CAS PubMed.
- J. R. Prensner and A. M. Chinnaiyan, Nat. Med., 2011, 17, 291–293 CrossRef CAS PubMed.
- F. Wu, H. Jiang, B. Zheng, M. Kogiso, Y. Yao, C. Zhou, X. N. Li and Y. Song, J. Med. Chem., 2015, 58, 6899–6908 CrossRef CAS PubMed.
- D. Rohle, J. Popovici-Muller, N. Palaskas, S. Turcan, C. Grommes, C. Campos, J. Tsoi, O. Clark, B. Oldrini, E. Komisopoulou, K. Kunii, A. Pedraza, S. Schalm, L. Silverman, A. Miller, F. Wang, H. Yang, Y. Chen, A. Kernytsky, M. K. Rosenblum, W. Liu, S. A. Biller, S. M. Su, C. W. Brennan, T. A. Chan, T. G. Graeber, K. E. Yen and I. K. Mellinghoff, Science, 2013, 340, 626–630 CrossRef CAS PubMed.
- B. Zheng, Y. Yao, Z. Liu, L. Deng, J. L. Anglin, H. Jiang, B. V. Prasad and Y. Song, ACS Med. Chem. Lett., 2013, 4, 542–546 CrossRef CAS PubMed.
- J. Popovici-Muller, J. O. Saunders, F. G. Salituro, J. M. Travins, S. Yan, F. Zhao, S. Gross, L. Dang, K. E. Yen, H. Yang, K. S. Straley, S. Jin, K. Kunii, V. R. Fantin, S. Zhang, Q. Pan, D. Shi, S. A. Biller and S. M. Su, ACS Med. Chem. Lett., 2012, 3, 850–855 CrossRef CAS PubMed.
- U. C. Okoye-Okafor, B. Bartholdy, J. Cartier, E. N. Gao, B. Pietrak, A. R. Rendina, C. Rominger, C. Quinn, A. Smallwood, K. J. Wiggall, A. J. Reif, S. J. Schmidt, H. Qi and H. Zhao, Nat. Chem. Biol., 2015, 11, 878–886 CrossRef CAS PubMed.
- G. Deng, J. Shen, M. Yin, J. McManus, M. Mathieu, P. Gee, T. He, C. Shi, O. Bedel, L. R. McLean, F. Le-Strat, Y. Zhang, J. P. Marquette, Q. Gao, B. Zhang, A. Rak, D. Hoffmann, E. Rooney, A. Vassort, W. Englaro, Y. Li, V. Patel, F. Adrian, S. Gross, D. Wiederschain, H. Cheng and S. Licht, J. Biol. Chem., 2015, 290, 762–774 CrossRef CAS PubMed.
- M. Niu, F. Dong, S. Tang, G. Fida, J. Qin, J. Qiu, K. Liu, W. Gao and Y. Gu, PLoS One, 2013, 8, e82360 Search PubMed.
- S. Zhang, J. Tan, Z. Lai, Y. Li, J. Pang, J. Xiao, Z. Huang, Y. Zhang, H. Ji and Y. Lai, J. Chem. Inf. Model., 2014, 54, 1785–1797 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- N. London, R. M. Miller, S. Krishnan, K. Uchida, J. J. Irwin, O. Eidam, L. Gibold, P. Cimermancic, R. Bonnet, B. K. Shoichet and J. Taunton, Nat. Chem. Biol., 2014, 10, 1066–1072 CrossRef CAS PubMed.
- C. Liu, G. He, Q. Jiang, B. Han and C. Peng, Int. J. Mol. Sci., 2013, 14, 14225–14239 CrossRef PubMed.
- S. Yao, T. Lu, Z. Zhou, H. Liu, H. Yuan, T. Ran, S. Lu, Y. Zhang, Z. Ke, J. Xu, X. Xiong and Y. Chen, Mol. Diversity, 2014, 18, 183–193 CrossRef CAS PubMed.
- Y. Chen, L. Fang, S. Peng, H. Liao, J. Lehmann and Y. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 3181–3187 CrossRef CAS PubMed.
- S. Chen, Y. Wang, W. Zhou, S. Li, J. Peng, Z. Shi, J. Hu, Y. C. Liu, H. Ding, Y. Lin, L. Li, S. Cheng, J. Liu, T. Lu, H. Jiang, B. Liu, M. Zheng and C. Luo, J. Med. Chem., 2014, 57, 9028–9041 CrossRef CAS PubMed.
- C. Zhou, D. Kang, Y. Xu, L. Zhang and X. Zha, Chem. Biol. Drug Des., 2015, 85, 659–671 CAS.
- J. L. Dahlin, J. W. Nissink, J. M. Strasser, S. Francis, L. Higgins, H. Zhou, Z. Zhang and M. A. Walters, J. Med. Chem., 2015, 58, 2091–2113 CrossRef CAS PubMed.
- S. Pusch, L. Schweizer, A. C. Beck, J. M. Lehmler, S. Weissert, J. Balss, A. K. Miller and A. von Deimling, Acta Neuropathol. Commun., 2014, 2, 19 CrossRef PubMed.
- J. Balss, S. Pusch, A. C. Beck, C. Herold-Mende, A. Kramer, C. Thiede, W. Buckel, C. D. Langhans, J. G. Okun and A. von Deimling, Acta Neuropathol., 2012, 124, 883–891 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21617j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |