DOI:
10.1039/C6RA12682K
(Paper)
RSC Adv., 2016,
6, 96743-96751
3-Dimensional cuboid structured ZnFe2O4@C nano-whiskers as anode materials for lithium-ion batteries based on the in situ graft polymerization method
Received
16th May 2016
, Accepted 16th September 2016
First published on 19th September 2016
Abstract
3-Dimensional cuboid structured ZnFe2O4@C nano-whiskers anode materials have been successfully synthesized via an in situ graft copolymerization method and the subsequent calcination process. Polystyrene-acrylonitrile (PSA) serves as the coating layer, which plays an important role in the calcination process. The final electrode materials were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The results of electrochemical tests demonstrate an excellent electrochemical performance, including good rate capability (over 700 mA h g−1 at the current density of 3.2 A g−1) and good cycling performance (a reversible capacity of 1722 mA h g−1 after 120 cycles with coulombic efficiency of 98.4%). Therefore, we believe that the proposed work may be a potential method to assist ZnFe2O4 to be a quite promising alternative anode material for lithium-ion batteries (LIBs).
1. Introduction
With the continuous development of science and technology and the growing commercial demand, more and more attention has been paid to various new energy storage means. In addition, severe ecological and environmental issues and energy shortages urgently need to be ameliorated.1 Among the multifarious new electrical energy storage solutions, LIBs, having the advantage of practicability as rechargeable batteries, greatly facilitate people's life and work. In recent years, LIBs have essentially been applied in various fields, such as small portable electronic equipment, pure and plug-in hybrid electric vehicles, power generation equipment and so on.2–6 The electrode material is the key factor that determines the overall performance of LIBs. Commercial graphite as the anode material of LIBs possesses the advantages of good reversibility, high capacity and low discharge plateau. However, the relatively low capacity of graphite (372 mA h g−1) has reached its theoretical limit, which cannot meet the ever-growing capacity requirements of high energy density consumption. Therefore, it is vital to develop new anode materials to satisfy the ongoing trend toward high performance LIBs.7–12
As anode materials, ferrite materials, especially the ternary oxide (MFe2O4, where M = Cu, Ni, Co, Zn, etc.) materials,13,14 have attracted significant attention, based on the advantages of their high capacity, relatively stable cyclability and abundance in nature.15–18 Among the various Fe-based oxides, ZnFe2O4 material stands out as a promising anode material, due to its relatively high theoretical capacity (about 1000 mA h g−1) resulting from Zn2+ reacting with Li+ to produce ZnLix alloy.19–25
The main challenge to the practical application of ZnFe2O4 is that their low electronic conductivity results in poor, high-rate charge–discharge properties, low actual specific capacity, large bulk variation during the Li-ion insertion and extraction processes, and the formation of a solid electrolyte interphase, namely SEI film, which lead to capacity recession in continuous cycling.26 To solve these problems, numerous research efforts have been implemented, including the urea combustion method, element doping, surface modification and morphology optimization, such as size control (to nanoscale), specific particulate morphology and the amelioration of the pore structure.27–30 For instance, the urea combustion method was used by Chowdari to synthesize ZnFe2O4, reporting a reversible capacity of 615 mA h g−1 after 50 cycles.31 Mn-doped ZnFe2O4 synthesized by Teh et al. achieved a reversible capacity up to 1417 mA h g−1 in the initial charge–discharge process and a specific capacity of 612 mA h g−1 at the current density of 65 mA g−1 after 100 cycles.32 With using a polymer pyrolysis method, the nano-structured ZnFe2O4 exhibited a capacity of more than 800 mA h g−1 at a specific current of 116 mA g−1 after 50 charge–discharge cycles.33 The nano-composite of ZnFe2O4 and graphene as the anode material achieved a capacity of 956 mA h g−1 at a current density of 100 mA g−1 after cycling 50 times, and porous, hollow ZnFe2O4 microspheres remained at 584 mA h g−1 after 100 cycles at 100 mA g−1 (ref. 21 and 34).
In this article, 3-D cuboid structured ZnFe2O4@C anode materials are successfully synthesized, based on a facile in situ graft copolymerization approach. This approach includes two steps, surface modification of ZnFe2O4 particles and monomer polymerization. Due to the strong surface polarity and high surface energy of the nano ZnFe2O4 material in the thermodynamically unsteady state, the nano-particles agglomerate easily. The water molecules in the air react easily with the surface of the nano ZnFe2O4 particles, on which many hydroxyl groups could form. The silane coupling agent, γ-methacryloxypropyltrimethoxysilane (KH570), is applied as a surface modifier for grafting active functional groups onto the nano ZnFe2O4 particles;35,36 it is conducive to the formation of chemical bonds between the modified ZnFe2O4 particles and the polymer matrix, for ensuring a durable and strong chemical junction between the polymer monomers and ZnFe2O4 particles.37,38 This novel ZnFe2O4@C composite consisting of plenty of homogeneous nano-particles, shows excellent electrochemical performance. The reversible capacity is found to be no less than 1700 mA h g−1, while the current density remains at 100 mA g−1 after 120 cycles. The novel 3-D cuboid structure can greatly increase the efficiency of lithium-ion storage and also partly relieve the volumetric expansion.39,40 Therefore, this method may be recommended as a novel and usable experimental means for the carbon-coated electrode materials of LIBs.21,41–43
2. Experimental
2.1 Material synthesis
All chemicals (Sigma Aldrich) used were commercially available and of analytical grade and were used without further purification. A typical synthesis based on a facile in situ graft copolymerization approach is simply illustrated in Scheme 1. In the preparation of the ZnFe2O4 precursor, cyclohexane (100 mL), ethylene glycol (16.5 mL), deionized water (16.5 mL), and 10.68 g of cetyltrimethylammonium bromide (CTAB)44 were added to a beaker and completely dissolved under strong stirring. Oxalic acid (10 g) was then added as a precipitating agent, and stirred gently for 1 h. Afterwards, ZnSO4·7H2O (1.15 g) and FeSO4·7H2O (2.23 g) were dissolved in the mixed solution, so that the molar ratio of ZnSO4·7H2O and FeSO4·7H2O was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The mixed solution was stirred at room temperature for 4 h. With washing and drying, the final yellow ZnFe2O4 powder was obtained. Graft copolymerization was then carried out as follows. The ZnFe2O4 powder (5 g) and aqueous ethanol solution were stirred together for some time until the ZnFe2O4 powder was uniformly dispersed in the aqueous ethanol. The γ-methacryloxypropyltrimethoxysilane (coupling agent KH570) ethanol solution was then added dropwise (0.25 g KH570 dissolved in absolute ethanol). The stirring was continued for 8 h for surface modification of the ZnFe2O4 nano-particles, which were covered by organic functional groups from KH570. Afterwards, the reaction system was heated to 60 °C, and a solution of ethanol and benzoyl peroxide (BPO) (0.05 g benzoyl peroxide was dissolved in absolute ethanol) as initiator was immediately poured into the system. The mixed monomer (1.5 g styrene (St) and 3.5 g acrylonitrile (AN)) and 2.3 g of polyvinylpyrrolidone (PVP) as the dispersant, with mass fraction of 25.18%, were completely dissolved in anhydrous ethanol and then dropped into the above reaction system. The in situ graft copolymerization was sustained at 60 °C for 12 h with continuous stirring. The final product was washed three times and dried at 60 °C for 8 h; subsequently, in a N2 atmosphere, the as-prepared precursor was sintered in a muffle furnace at 500 °C for 5 h to obtain a carbon-coated ZnFe2O4 powder.
:2. The mixed solution was stirred at room temperature for 4 h. With washing and drying, the final yellow ZnFe2O4 powder was obtained. Graft copolymerization was then carried out as follows. The ZnFe2O4 powder (5 g) and aqueous ethanol solution were stirred together for some time until the ZnFe2O4 powder was uniformly dispersed in the aqueous ethanol. The γ-methacryloxypropyltrimethoxysilane (coupling agent KH570) ethanol solution was then added dropwise (0.25 g KH570 dissolved in absolute ethanol). The stirring was continued for 8 h for surface modification of the ZnFe2O4 nano-particles, which were covered by organic functional groups from KH570. Afterwards, the reaction system was heated to 60 °C, and a solution of ethanol and benzoyl peroxide (BPO) (0.05 g benzoyl peroxide was dissolved in absolute ethanol) as initiator was immediately poured into the system. The mixed monomer (1.5 g styrene (St) and 3.5 g acrylonitrile (AN)) and 2.3 g of polyvinylpyrrolidone (PVP) as the dispersant, with mass fraction of 25.18%, were completely dissolved in anhydrous ethanol and then dropped into the above reaction system. The in situ graft copolymerization was sustained at 60 °C for 12 h with continuous stirring. The final product was washed three times and dried at 60 °C for 8 h; subsequently, in a N2 atmosphere, the as-prepared precursor was sintered in a muffle furnace at 500 °C for 5 h to obtain a carbon-coated ZnFe2O4 powder.
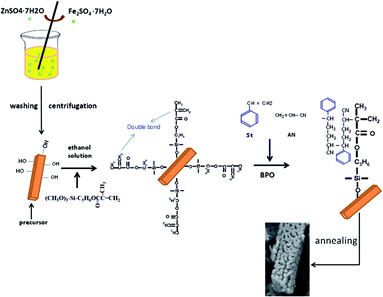 |
| | Scheme 1 Schematic illustration of the synthesis procedure for the anode nanoparticles. | |
2.2 Material characterization
The crystallographic texture of the composite was investigated by X-ray diffraction (XRD, PANalytical X'pert PRO, Cu/Kα radiation, λ = 0.15406 nm). Surface analysis of the sample was carried out using X-ray photoelectron spectroscopy (XPS, ESCALAB 250 with a 150 W Al Kα probe beam). The morphology and microstructure of the samples were examined by scanning electron microscope (SEM, ZEISSULTRA55) and transmission electron microscope (TEM, JEM-2100HR). Raman spectra were acquired with a HR800UV Raman microspectrometer (Jobin Yvon LabRam) to further demonstrate the existence of a carbon layer in the final prepared composite powders. The pore size distribution and mesoporous structure were determined using Barrett Joyner Halenda (BJH) analysis (NO-VA3200e, Quanta chrome).
2.3 Electrochemical measurement
The electrochemical experiment was conducted using CR2530 coin-type cells, which were assembled with lithium foil as the counter electrode in an argon-filled glove box. The working electrodes contained paste-like active materials, super-P and PVDF, in a ratio of 8:1:1 distribution. The above substances were mixed with a high-speed stirring method for several hours to produce a smooth viscous fluid. The slurry was then evenly coated on copper (Cu) foil. The coating mass on the Cu foil was 1.5 mg on average. Super P, PVDF and the copper foil were received from BTR Battery Materials Co. Ltd. After drying for 8 h at 80 °C, the electrode was cut into a circle of 18 mm in diameter, and the electrolyte was 1.0 M LiPF6 in a mixture of DEC, EC, EMC with volume ratio of 1:1:1 (afforded by Chei Industries Inc., South Korea). A Celgard 2400 membrane was used as a separator. In addition to the experimental sample, the pure ZnFe2O4 served as the active material for the blank controlled group. Galvanostatic charge–discharge tests were performed at a current density of 100 mA g−1 in the voltage limits of 0.01 V–3.0 V vs. Li+/Li. Cyclic voltammetry (CV) was carried out with a Solartron Analytical 1470E electrochemistry system at a scanning rate of 0.2 mV s−1. All electrochemical measurements were conducted by a LAND CT2001A battery testing system at 25 °C.
3. Results and discussion
3.1 Phase and microstructures
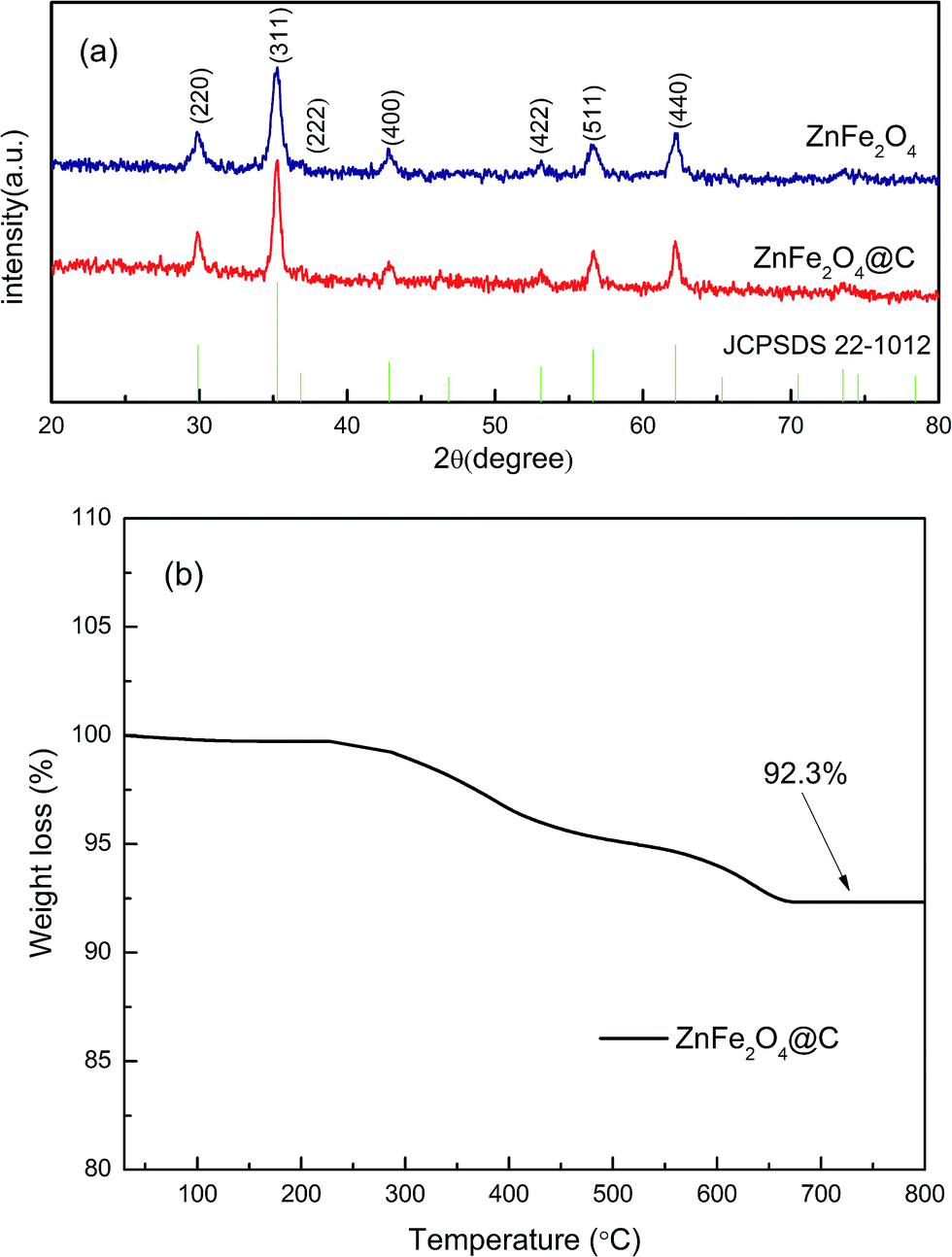
The XRD patterns of the obtained products are recorded in the 2θ range, 20–80°, as shown in Fig. 1. The XRD pattern of 3D cuboid structured ZnFe2O4@C is consistent with spinel ZnFe2O4 (JCPDS Card No. 89-1010). The diffraction peaks, located at 2θ of 30.1°, 35.72°, 37.06°, 42.98°, 53.33°, 56.79°, and 62.41°, correspond well to the (220), (311), (222), (400), (422), (511), and (440) planes, respectively, of the spinel-type ZnFe2O4 crystal phase. No additional peak was found, suggesting that no by-products, such as ZnO, were produced, indicating that the final products have high purity and excellent crystallinity. Compared to the spectra of pure ZnFe2O4, the relatively narrower and stronger peaks of ZnFe2O4@C may be attributed to polystyrene-acrylonitrile (PSA) as carbonaceous matrixes, increasing the crystallinity of calcined samples. No obvious peak corresponding to carbon was found in the XRD pattern of the 3D cuboid structured ZnFe2O4@C material, indicating that the carbon in the sample is amorphous, as confirmed by Raman spectra.
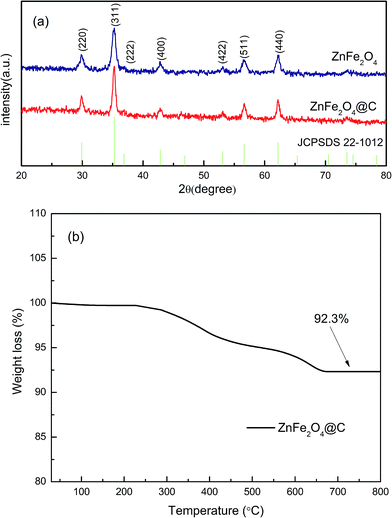 |
| | Fig. 1 (a) XRD patterns of the ZnFe2O4 powder coated with and without carbon, (b) the TGA curve of ZnFe2O4@C. | |
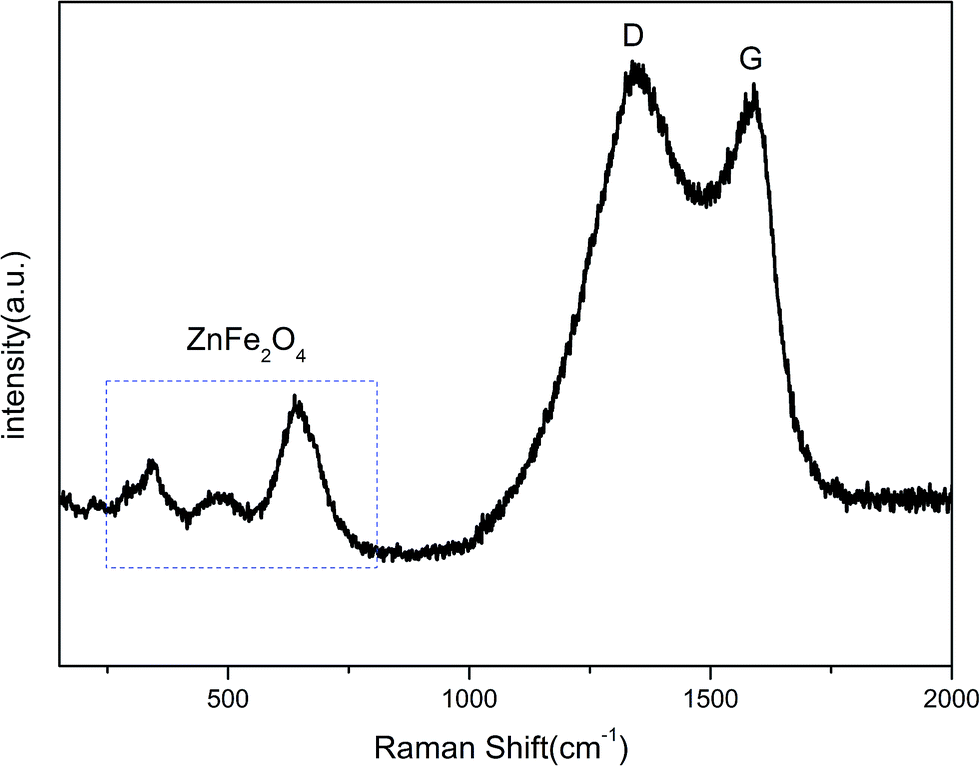
Since the XRD pattern cannot judge whether the ZnFe2O4 was coated with carbon after annealing, TG testing was applied. Fig. 1(b) shows the TG curve of ZnFe2O4@C nano-composites under air atmosphere in the range of 30–800 °C. According to the mass loss curve, the carbon content in the ZnFe2O4@C composite was derived to be approximately 7.7 wt%. Raman spectra were further used to determine the existence of amorphous carbon. In Fig. 2, two obvious adsorption peaks are viewed at 1353.82 and 1589.52 cm−1, corresponding to D and G bands, respectively. The D and G bands refer to the sp2 bond stretching vibration mode. The relative strength of the D band is a reflection of the degree of crystal structural disorder, while the G band represents the first order scattering with the E2g vibration mode, which is used to characterize the structure of the carbon sp2 bond.45 The intensity ratio of ID/IG of ZnFe2O4@C was calculated to be 1.03, which indicates a great degree of atomic order.46 This obviously confirms the presence of carbon in the final product corresponding to the TG curve in Fig. 1(b). Besides, three characteristic peaks in the range of 200–1000 cm−1 agree well with previously reported work on ZnFe2O4 nanoparticles.29
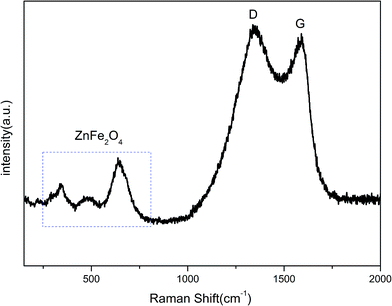 |
| | Fig. 2 Raman spectrum of ZnFe2O4@C. | |
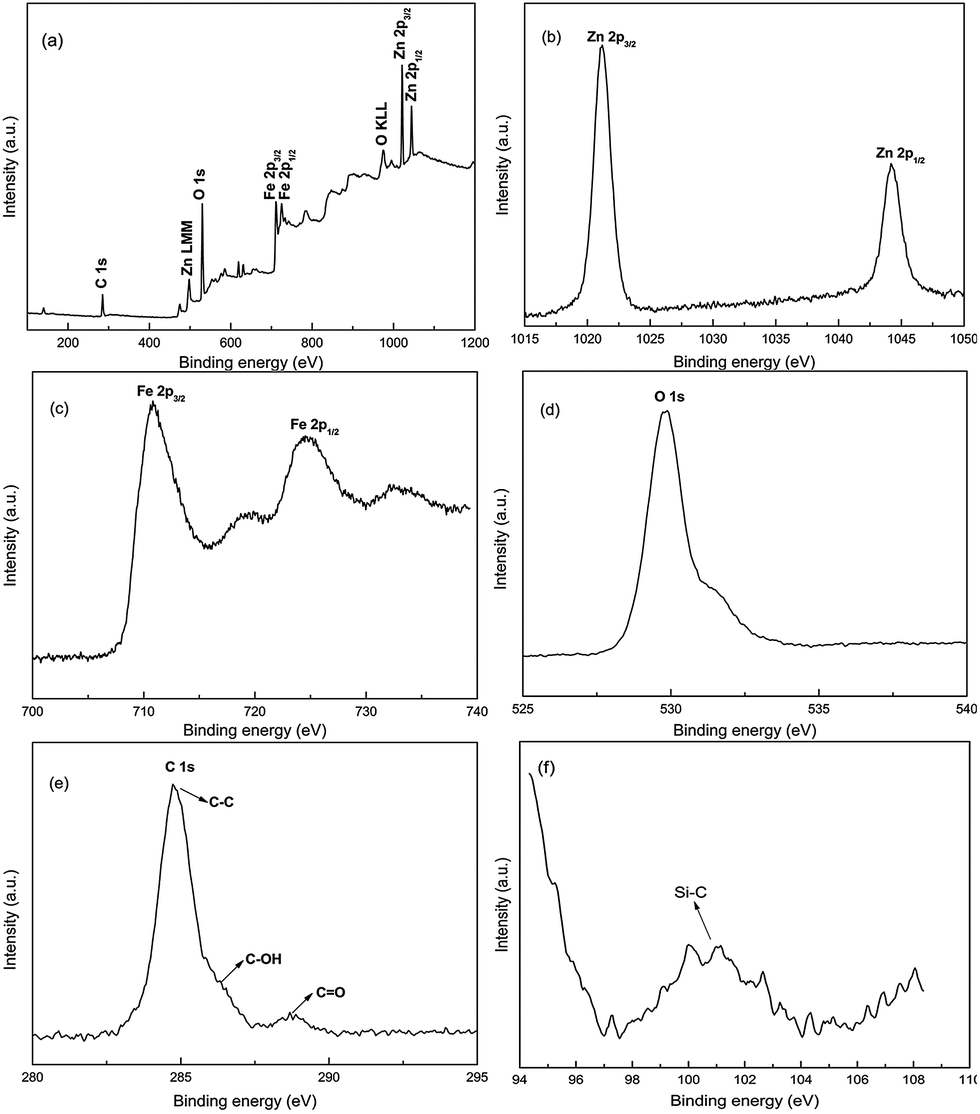
To elucidate the oxidation states of chemical elements and the surface composition of ZnFe2O4@C nanocomposites, the full-scale XPS survey spectra in the region of 100–1200 eV are shown in Fig. 3, confirming the existence of Zn, Fe, O, C. The peaks at binding energies of 1021.18 eV and 1044.18 eV could be described as Zn 2p3/2 and Zn 2p1/2,47 respectively, which indicate that the valence state of Zn is +2 in the final sample. In Fig. 3(b), the two main peaks observed at binding energies of 710.68 eV and 724.53 eV correspond well to Fe3+ at octahedral sites,48 while the two satellite peaks observed at binding energies of 719.88 eV and 732.13 eV correspond to the +3 oxidation status of iron. A strong peak is shown in Fig. 3(d) at the binding energy of 529.78 eV, which is ascribed to the O2− in metallic oxide. In Fig. 3(e), a sharp peak at the binding energy of 284.73 eV indicates that the polymer is well anchored to the ZnFe2O4 nanoparticles during the polymerization process. The two weak peaks at the binding energies of 286.53 eV and 288.68 eV correspond to C–OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups, respectively, which implies that the copolymer is well combined with the oxygen-containing functional groups on the surface of ZnFe2O4 nanoparticles after sintering.43 From the testing result in Fig. 3(f), the major peak is located at the binding energy of ∼101 eV; this is not the peak position of the silicon–oxygen bond. The location of the peak is at the binding energy of ∼101 eV, corresponding to the silicon–carbon functional groups. Besides, according to the structural formula of KH570, the silicon–oxygen bond breaks easily in aqueous solution, for binding with hydroxyl groups, which is in keeping with our experiment, where the γ-methacryloxypropyltrimethoxysilane (coupling agent KH570) occupied a very small proportion. It has almost no impact on our experiment data.
O functional groups, respectively, which implies that the copolymer is well combined with the oxygen-containing functional groups on the surface of ZnFe2O4 nanoparticles after sintering.43 From the testing result in Fig. 3(f), the major peak is located at the binding energy of ∼101 eV; this is not the peak position of the silicon–oxygen bond. The location of the peak is at the binding energy of ∼101 eV, corresponding to the silicon–carbon functional groups. Besides, according to the structural formula of KH570, the silicon–oxygen bond breaks easily in aqueous solution, for binding with hydroxyl groups, which is in keeping with our experiment, where the γ-methacryloxypropyltrimethoxysilane (coupling agent KH570) occupied a very small proportion. It has almost no impact on our experiment data.
 |
| | Fig. 3 XPS spectra of ZnFe2O4@C nanocomposites: (a) survey spectrum, (b) Zn 2p, (c) Fe 2p, (d) O 1s, (e) C 1s, (f) Si–C. | |
The micromechanism and morphologies of pure ZnFe2O4 and the ZnFe2O4@C were tested by SEM, as shown in Fig. 4. The pure ZnFe2O4 (Fig. 3(a)) exhibits a smoother cuboid shape and the length mostly ranges between 3 μm and 5 μm. Fig. 4(b–d) show that the ZnFe2O4@C is composed of numerous small particles, evenly distributed on long rectangles akin to the 3-D cuboid structure. The average particle size is ∼50 nm. It can be seen that the arrangement of superficial particles is uniform and flat. Moreover, there are plenty of mesoporous structures among the nanoparticles. This structure not only alleviates the huge volume expansion effect during lithium-ion insertion and extraction processes, and increases the capacity of lithium-ion insertion, but also it shortens the transmission distance of the lithium-ion. A large number of nano-particles with huge superficial area provide a tremendous contact area with the electrolyte that promotes the lithium-ion diffusion and electron transport during cycling. Such voids could facilitate the diffusion and penetration of liquid electrolyte, thus contributing to the rate performance.49
 |
| | Fig. 4 SEM images of (a) the pure ZnFe2O4 and (b–d) the 3-dimensional cuboid ZnFe2O4@C with different magnifications. | |
TEM was employed to obtain more information related to the morphology and microstructure of the as-prepared ZnFe2O4@C and pure ZnFe2O4. Fig. 5(a), (b), (e) and (f) show that the materials are both composed of numerous nanoparticles, and a carbon layer can be clearly seen on the surface in Fig. 5(a) and (b), while there are no layers in Fig. 5(e) and (f). The carbon layer is also demonstrated in Fig. 5(c), marked using black dash lines. In Fig. 5(c) and (g), visible parallel lattice fringes can be observed. The interplanar spacing between two adjacent lattice fringes is 0.307 nm and 0.305 nm, respectively, corresponding well to the (511) plane of the crystalline ZnFe2O4 phase (JCPDS: 821049). Fig. 5(d) and (h) show the electron diffraction patterns that contain many of the same clear rings, according to the (220) (311) (400), and (511) planes of ZnFe2O4, which are consistent with the polycrystalline structure and the XRD results.
 |
| | Fig. 5 TEM images of (a), (b) ZnFe2O4@C cuboids and (e), (f) ZnFe2O4 cuboids; HR-TEM images of (c) ZnFe2O4@C cuboids and (g) ZnFe2O4 cuboids; and the SAED patterns of (d) ZnFe2O4@C and (h) ZnFe2O4 cuboids. | |
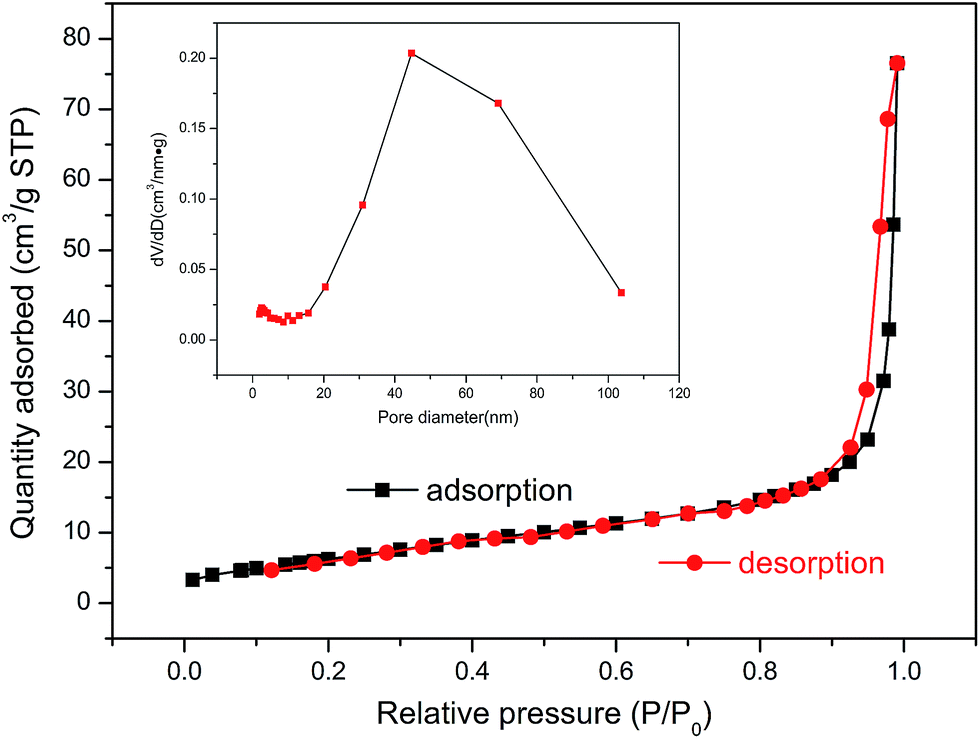
In Fig. 6, the nitrogen adsorption/desorption isotherms of ZnFe2O4@C belong to the type IV category according to the International Union of Pure and Applied Chemistry classification, suggesting the presence of a mesoporous structure, owing to the distinct hysteresis loop between P/P0 of 0.9–1.0. The inset in Fig. 6 shows a pore size distribution curve calculated from the adsorption branch of isotherms, which is subjected to Barrett Joyner Halenda (BJH) measurement. It illustrates that the pore size is mainly located in the range of 40–80 nm, while the peak is at around 45 nm. The distribution of pore size is broadly in line with the SEM images, as shown in Fig. 4(c) and (d). The Brunauer–Emmett–Teller (BET) specific surface area of ZnFe2O4@C was determined to be 24.3 m2 g−1, which shows that a relatively void space exists among the self-assembled nanoparticles. This would potentially contribute to preventing the phenomenon of the nullity of the lithium-ion. Typically, the increase in particle size on account of sintering would lead to a porous structure, since the pores are formed from the stacking of a mass of nano-particles. It suggests that the carbon formed from the carbonization of copolymer for the synthesis of ZnFe2O4@C acts as a diffusion block to effectively suppress the growth of ZnFe2O4 nanoparticles during the calcination process.
 |
| | Fig. 6 Nitrogen adsorption/desorption isotherms of the ZnFe2O4@C, and the pore-size distribution (inset) analyzed by the Barrett Joyner Halenda (BJH) method. | |
3.2 Electrochemical performance
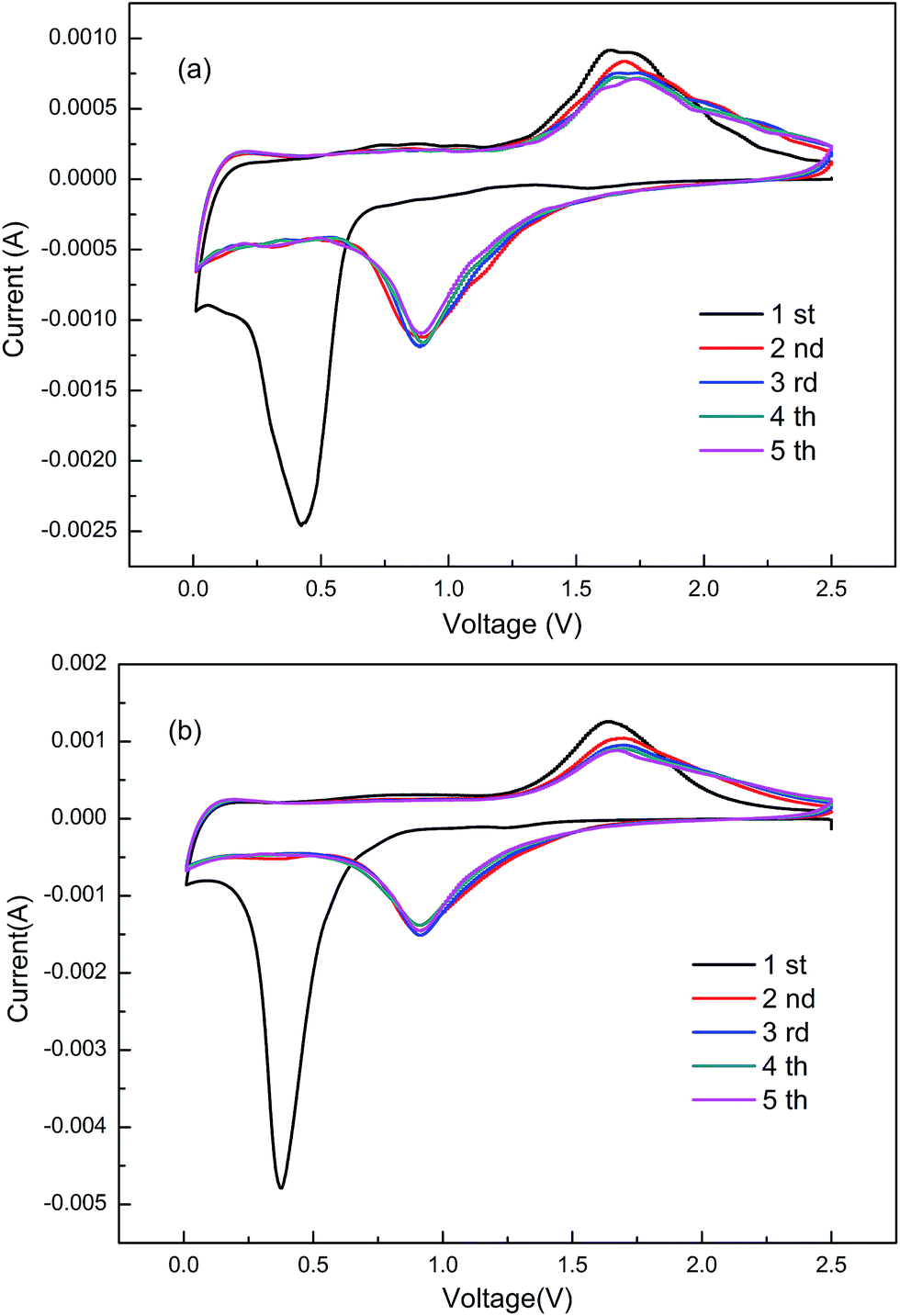
In Fig. 7, the cyclic voltammetry (CV) curves are used to record the thermodynamics of the lithiation process in pure ZnFe2O4 and ZnFe2O4@C electrodes at a scanning rate of 0.2 mV s−1 with the potential range from 2.5 V to 0.01 V from the 1st to 5th cycle. The two groups of curves have similar locations of the reductive peak and oxidation peak at voltages of ∼0.4 V and ∼1.65 V in the 1st cycle, which illustrate the same reaction potential of the two electrodes. In the first cathodic process, only an obvious sharp narrow reduction peak appears at ∼0.4 V, which is ascribed to the reduction reaction of ZnFe2O4 with Fe3+, Fe2+, Zn2+ to give Fe0, Zn0, the formation of Li–Zn alloys, and the generation of solid electrolyte interphase (SEI) film, due to the decomposition of the electrolyte. In the anodic process, the narrow peak in the first cycle located at 1.65 V is attributed to the oxidation of Zn and Fe to Zn2+and Fe3+, respectively.50 In the following cycles, the sharp peak no longer appears in both cathodic and anodic processes. This phenomenon implies that the irreversible capacity loss is still inevitable in the electrochemical cycling because of the electrode pulverization and the formation of SEI film. Nevertheless, compared to the pure ZnFe2O4 in Fig. 7(a), all the characteristic peaks and integrated areas of ZnFe2O4@C in Fig. 7(b) remain unchanged in the next four cycles, which suggests good cycling stability of the ZnFe2O4@C electrode.
 |
| | Fig. 7 Cyclic voltammetry curves of (a) the as-obtained pure ZnFe2O4 electrode and (b) ZnFe2O4@C electrode. | |
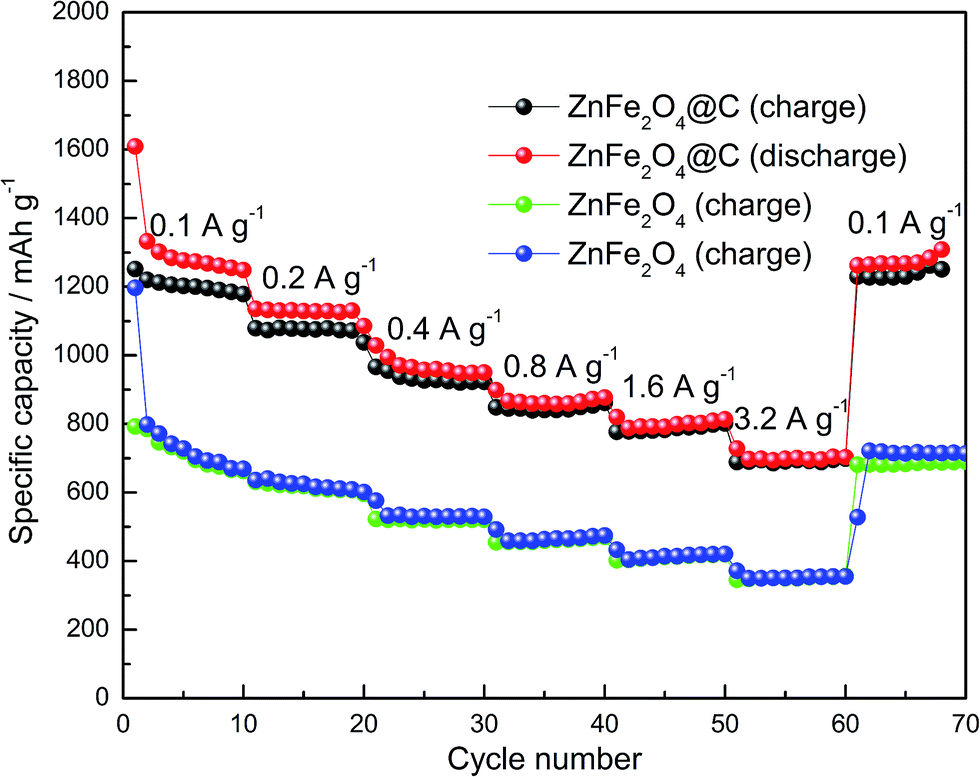
To compare the electrochemical performance of the ZnFe2O4@C composite and pure ZnFe2O4, the rate capacity was tested, as shown in Fig. 8. The initial discharge and charge capacity of the ZnFe2O4@C electrode reached 1609 mA h g−1 and 1251 mA h g−1, respectively. The large irreversible capacity loss may have resulted from the formation of SEI film in the electrode. Different current densities during 0.1–3.2 A g−1 were applied for the experiments. The current magnitude doubled every ten charge–discharge cycles. It can be seen that the discharge–charge capacity still remains as high as over 700 mA h g−1 at the current density of 3.2 A g−1, suggesting a satisfactory rate capacity of the electrode. Moreover, the reversible capacity can attain over 1200 mA h g−1 at 100 m A g−1 after 70 cycles. In contrast, the rate capacity of pure ZnFe2O4 electrode is apparently lower than that of the ZnFe2O4@C electrode at various current densities, including the first cycle. It is found to be attenuated in high rate charge–discharge processes, such as at the current density of 1.6 A g−1 and 3.2 A g−1. After 70 charge–discharge cycle times, the reversible capacity is lower than the capacity in the first cycling. The superior electrochemical performance of the ZnFe2O4@C electrode could be ascribed to the 3-D cuboid nanostructure and the strong bonds between the precursor and carbon after polymer carbonization, which not only acts as the buffer layer to maintain the structural stability, but also increases the electrical conductivity during the charge–discharge processes.
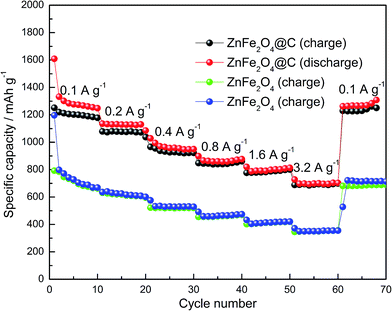 |
| | Fig. 8 Rate capability of the ZnFe2O4@C and pure ZnFe2O4 electrodes. | |
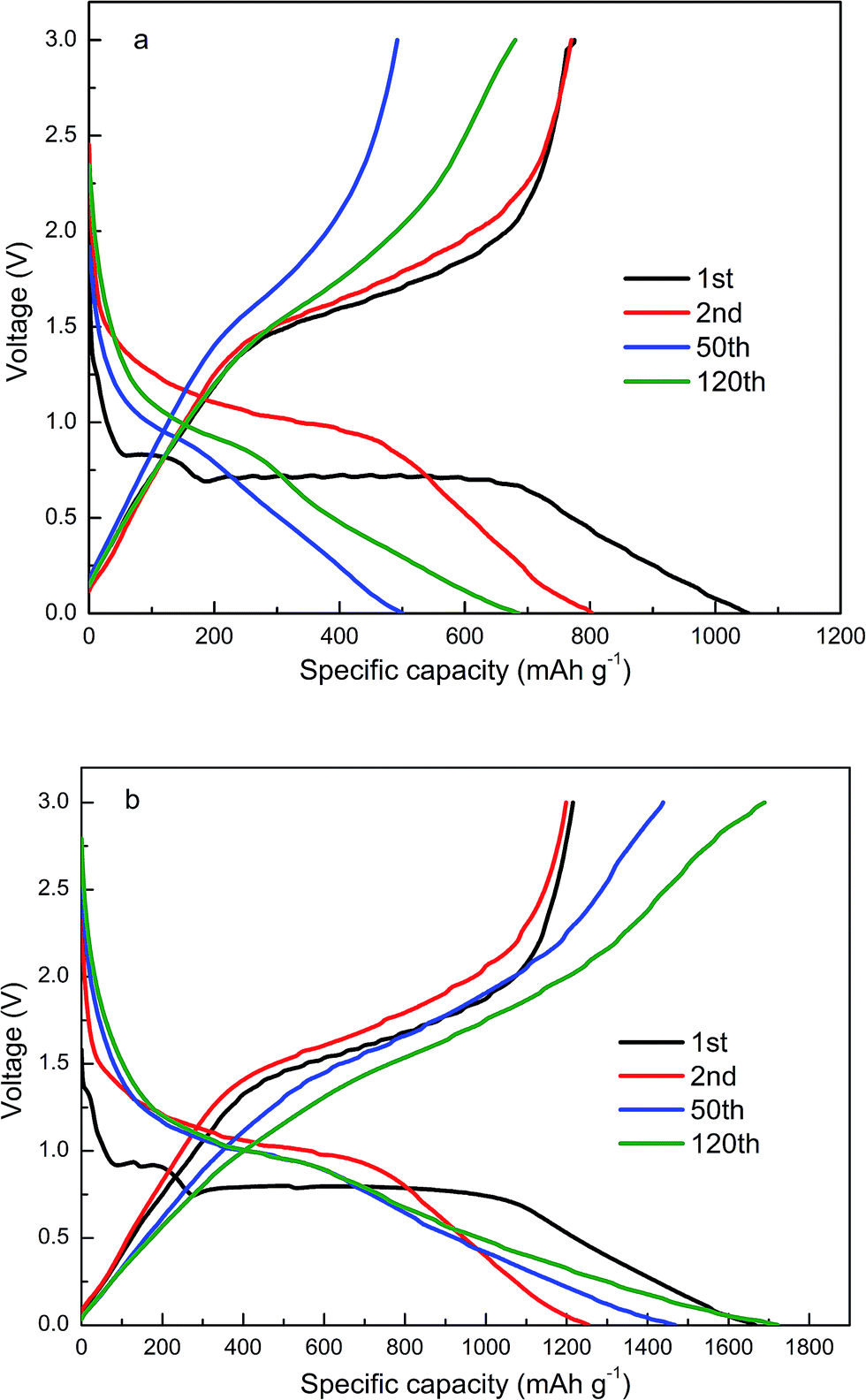
The Fig. 9(a) and (b) clearly show the discharge/charge curves at the current density of 100 mA g−1 of the pure ZnFe2O4 and ZnFe2O4@C within a potential window of 0.01–3.0 V. Two obvious voltage plateaus of the 1st discharge profile at approximately 0.9 and 0.8 V are shown in Fig. 9(b). The shorter plateau at 0.9 V exhibits Li+ insertion into the ZnFe2O4 crystal lattice. The longer plateau at 0.8 V suggests that the generated Li2ZnFe2O4 reacts with Li+ to form metallic Zn and Fe and the formation of Li2O. The discharge capacity in Fig. 9(b) reaches 1667 mA h g−1, 1254 mA h g−1, 1468 mA h g−1 and 1722 mA h g−1 at the 1st, 2nd, 50th and 120th cycles, respectively, which are much higher than those of Fig. 9(a). The capacity of pure ZnFe2O4 decayed much faster than the ZnFe2O4@C electrode. The initial irreversible capacity may be associated with the formation of a rather thick SEI film, which consumed more Li+ and irreversibly and partially embedded it. After the 1st cycle, the plateau gradually disappeared and the special capacity still reached up to 1722 mA h g−1 at the 120th cycle, which indicates that the electrode materials have a good reversibility. In addition, the reaction mechanism of Li+ insertion and extraction with ZnFe2O4 is displayed as follows:51
| | |
ZnFe2O4 + 0.5Li+ + 0.5e− → Li0.5ZnFe2O4
| (1) |
| | |
Li0.5ZnFe2O4 + 1.5Li+ + 1.5e− → Li2ZnFe2O4
| (2) |
| | |
Li2ZnFe2O4 + 6Li+ + 6e− → 4Li2O + Zn + 2Fe
| (3) |
| | |
3Li2O + 2Fe → Fe2O3 + 6Li+ + 6e−
| (5) |
| | |
Li2O + Zn → ZnO + 2Li+ + 2e−
| (6) |
 |
| | Fig. 9 Discharge–charge profiles of the (a) pure ZnFe2O4 and (b) ZnFe2O4@C for the 1st, 2nd, 50th and 120th cycle at the specific current of 100 mA g−1. | |
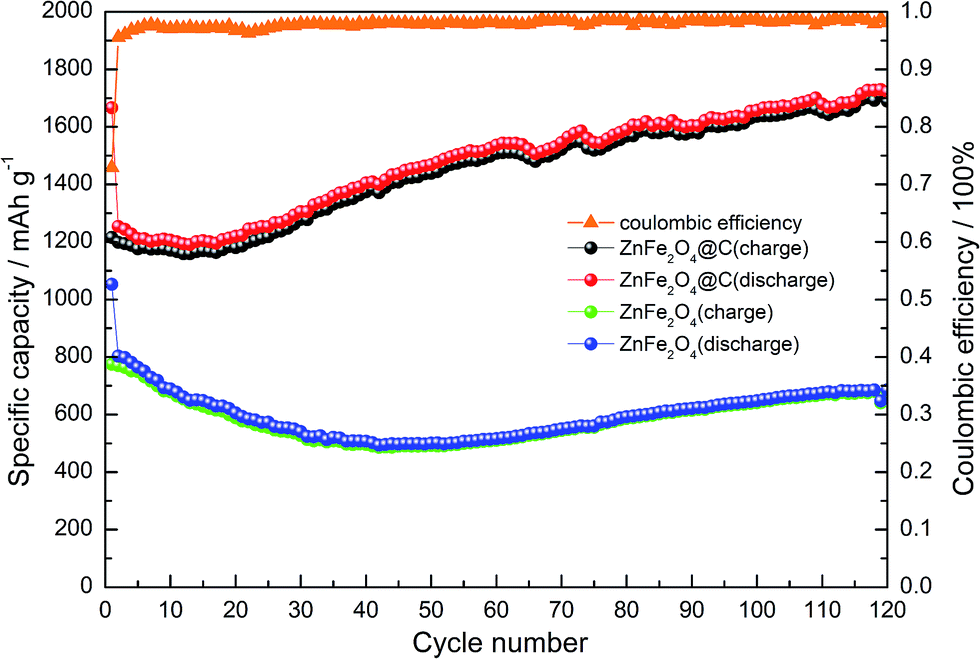
As is known, the power performance of LIBs is greatly limited by the slow diffusion of lithium-ions across the two phase boundary and the slow electronic conductivity.52 Fig. 10 shows the cycling performance and the coulombic efficiency of the pure ZnFe2O4 and anode ZnFe2O4@C hybrid anode at a constant current density of 100 mA g−1 for 120 cycles. Although the capacity retention in the first cycle is not as high as expected, it still quickly increased to over 98% in the second cycle and then remains high in the following cycles, which indicates a good reversibility during the lithium insertion and extraction process of the ZnFe2O4@C composite. What is more, for the pure ZnFe2O4 electrode, the specific capacity decreases from the 1st cycle to the 40th cycle, while the ZnFe2O4@C electrode only has a slight decline in the first 15 cycles. The specific capacity of pure ZnFe2O4 electrode had a small increase, while the specific capacity of ZnFe2O4@C electrode increased over 400 mA h g−1 in the 120th cycle relative to the first cycle. The higher increased specific capacity is likely to be ascribed to the material's activation after the initial charge–discharge process, with the help of the constant infiltration of the electrolyte, and Li+ reacted with more active materials that were not reachable at first. Due to the graft copolymerization, substantial carbon bonds between the surface of the ZnFe2O4 particles and copolymer protected the structure from collapsing easily and buffered the volume change of ZnFe2O4 during the process of Li+ insertion/extraction. In addition, during the charge and discharge process, the moderate pulverization of active materials would form many interfaces that probably offer more lithium-ion storage sites. Then, in the electrode, the nanosize effect of ZnFe2O4@C would have some assistance for the decomposition of Li2O, which would contribute to a part of the specific capacity.53 The surface of numerous nanoparticles can not only carry Li-ions, but can also carry charge, so that capacitance effects might also provide capacity contribution. The coulombic efficiency, as high as ∼98%, demonstrated the superior cycling stability of the ZnFe2O4@C electrode materials. Compared to the pure ZnFe2O4, the better performance of ZnFe2O4@C may be explained by the unique morphology and nano-structure reconfiguration during the charge and discharge cycles, thus allowing the Li+ to get an unblocked path during the insertion and extraction process.
 |
| | Fig. 10 Cycling performance of the ZnFe2O4@C and pure ZnFe2O4 electrodes at a current density of 100 mA g−1. | |
To further investigate the electrochemical behavior of ZnFe2O4@C, the morphologies of the original and cycled electrodes are shown in Fig. 10. The button cell tested in Fig. 11(b) was cycled for 20 cycles at 100 mA g−1. Comparing Fig. 11(a) and (b), the morphologies are clearly different. However, the obvious 3-D cuboid structure of the active material could still be observed, indicating that the structure could be retained.
 |
| | Fig. 11 SEM image of ZnFe2O4@C electrode before cycling (a) and after 20 cycles (b). | |
4. Conclusions
The results of various characterizations, including the electrochemical performance, reveal that the in situ graft polymerization is a better approach to improving the defects of ZnFe2O4 as an active material for greatly enhancing the electrochemical properties of LIBs. As the anode material, the final ZnFe2O4@C composite exhibits a good reversible capacity with a coulombic efficiency of approximately 73% in the first cycle and excellent stability of high rate capability in each of the subsequent charge–discharge processes, with a coulombic efficiency of about 98%. The cycling performance shows an initial discharge–charge capacity of 1667 mA h g−1 and 1216 mA h g−1. Moreover, the specific capacity is maintained at 1722 mA h g−1 after 120 cycles with galvanostatic discharge–charge cycling at a current density of 100 mA g−1. Besides, the cycle curves show an increasing trend with a very stable discharge–charge process of the ZnFe2O4@C electrode. The particular construction and morphology formed from grafting copolymerization offer a very efficient path for lithium-ion diffusion, and protects the integrity of the structure with adequate space to bear the significant volume change during the Li+ insertion and extraction process, contributing to a better electrochemical Li-storage behavior. Consequently, this strategy may be a good method for synthesising other anode materials for LIBs.
Acknowledgements
This work was financially supported by the Scientific and Technological Plan of Guangdong Province (2016A050503040, 2016B010114002), The Scientific and Technological Plan of Guangzhou City (20160710322), the Hong Kong Polytechnic University (4-ZZDC and 1-ZVGH) and Strategic Plan (1-ZVCG).
Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- B. Lim, J. Jin, J. Yoo, S. Y. Han, K. Kim, S. Kang, N. Park, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2014, 50, 7723 RSC.
- Y. L. Xiao, J. T. Zai, X. M. Li, Y. Gong, B. Li, Q. Y. Han and X. F. Qian, Nano Energy, 2014, 6, 51–58 CrossRef CAS.
- G. N. Zhu, Y. G. Wang and Y. Y. Xia, Energy Environ. Sci., 2012, 5, 6652 CAS.
- A. S. Hameed, H. Bahiraei, M. V. Reddy, M. Z. Shoushtari, J. J. Vittal, C. K. Ong and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2014, 6, 10744–10753 CAS.
- L. M. Yao, X. H. Hou, S. J. Hu, J. Wang, M. Li, C. Su, M. O. Tade, Z. P. Shao and X. Liu, J. Power Sources, 2014, 258, 305 CrossRef CAS.
- Z. L. Zhang, Y. H. Wang, D. Li, Q. Q. Tan, Y. F. Chen, Z. Y. Zhong and F. B. Su, Ind. Eng. Chem. Res., 2013, 52, 14906–14912 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- G. Q. Zhang and X. W. D. Lou, Sci. Rep., 2013, 3, 1470 Search PubMed.
- Y. H. Xu, G. Q. Jian, Y. H. Liu, Y. J. Zhu, M. R. Zachariah and C. S. Wang, Nano Energy, 2014, 3, 26–33 CrossRef CAS.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513–523 CrossRef CAS.
- L. R. Hou, L. Lian, L. H. Zhang, G. Pang, C. Z. Yuan and X. G. Zhang, Adv. Funct. Mater., 2015, 25, 238 CrossRef CAS.
- Y. H. Chao, Y. Y. Huang, J. Y. Chang, S. H. Peng, W. Y. Tu, Y. J. Cheng, J. H. Hou and C. S. Hsu, J. Mater. Chem. A, 2015, 3, 20382–20388 CAS.
- R. M. Thankachan, M. M. Rahman, I. Sultana, A. M. Glushenkov, S. Thomas, N. Kalarikkal and Y. Chen, J. Power Sources, 2015, 282, 462 CrossRef CAS.
- P. R. Kumar and S. Mitra, RSC Adv., 2013, 3, 25058 RSC.
- X. Chen, K. M. Unruh, C. Y. Ni, B. Ali, Z. C. Sun, Q. Lu, J. deitzel and J. Q. Xiao, J. Phys. Chem. C, 2011, 115, 373–378 CAS.
- L. M. Yao, X. H. Hou, S. J. Hu, X. Q. Tang, X. Liu and Q. Ru, J. Alloys Comp., 2014, 585, 398–403 CrossRef CAS.
- P. F. Teh, Y. Sharma, S. S. Pramana and M. Srinivasan, J. Mater. Chem., 2011, 21, 14999 RSC.
- Y. Ding, Y. F. Yang and H. X. Shao, Electrochim. Acta, 2011, 56, 9433 CrossRef CAS.
- Y. F. Deng, Q. M. Zhang, S. D. Tang, L. T. Zhang, S. N. Deng, Z. C. Shi and G. H. Chen, Chem. Commun., 2011, 47, 6828–6830 RSC.
- F. Mueller, D. Bresser, E. Paillard, M. Winter and S. Passerini, J. Power Sources, 2013, 236, 87–94 CrossRef CAS.
- J. W. Mao, X. H. Hou, X. Y. Wang, G. N. He, Z. P. Shao and S. J. Hu, RSC Adv., 2015, 5, 31807 RSC.
- N. Wang, H. Xu, L. Chen, X. Gu, J. Yang and Y. T. Qian, J. Power Sources, 2014, 247, 163–169 CrossRef CAS.
- Z. Wang, Z. Wang, W. Liu, W. Xiao and X. W. Lou, Energy Environ. Sci., 2013, 6, 87 CAS.
- J. Jiang, Y. Li, J. Liu and X. Huang, Nanoscale, 2011, 3, 45–48 RSC.
- P. Wang, M. X. Gao, H. Pan, J. L. Zhang, C. Liang, J. H. Wang, P. Zhou and Y. F. Liu, J. Power Sources, 2013, 239, 466–474 CrossRef CAS.
- Z. P. Zeng, H. L. Zhao, J. Wang, P. P. Lv, T. H. Zhang and Q. Xia, J. Power Sources, 2014, 248, 15–21 CrossRef CAS.
- X. H. Hou, X. Y. Wang, L. M. Yao, S. J. Hu, Y. P. Wu and X. Liu, New J. Chem., 2015, 39, 1943–1952 RSC.
- M. Akram and M. Anis-ur-Rehman, J. Electron. Mater., 2014, 2, 485–492 CrossRef.
- Y. Sharma, N. Sharma, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2008, 53, 2380 CrossRef CAS.
- P. F. Teh, Y. Sharma, Y. W. Ko, S. S. Pramana and S. Madhavi, RSC Adv., 2013, 3, 2812–2821 RSC.
- Y. Ding, Y. F. Yang and H. X. Shao, Electrochim. Acta, 2011, 56, 9433–9438 CrossRef CAS.
- Z. Xing, Z. C. Ju, J. Yang, H. Y. Xu and Y. T. Qian, Nano Res., 2012, 5, 477 CrossRef CAS.
- J. Zhao, M. Milanov, M. M. C. G. Warmoeskerkena and V. Dutschk, Colloids Surf., A, 2012, 413, 273 CrossRef CAS.
- M. Sabzi, S. M. Mirabedini, J. Zohuriaan-Mehr and M. Atai, Prog. Org. Coat., 2009, 65, 222 CrossRef CAS.
- X. M. Xu, B. J. Li, H. M. Lu, Z. J. Zhang and H. G. Wang, Appl. Surf. Sci., 2007, 254, 1456 CrossRef CAS.
- J. Lin, J. A. Siddiqui and R. M. Ottenbrite, Polym. Adv. Technol., 2001, 12, 286 CrossRef.
- H. J. Kim, H. G. Jung, B. Scrosati and Y. K. Sun, J. Power Sources, 2012, 203, 115–120 CrossRef CAS.
- W. L. Zhang, X. H. Hou, J. D. Shen, S. J. Hu, Q. Ru and K. Lam, Electrochim. Acta, 2016, 188, 734–743 CrossRef CAS.
- D. Li, X. Li, S. Wang, Y. Zheng, L. Qiao and D. He, ACS Appl. Mater. Interfaces, 2014, 6, 648–654 CAS.
- F. Yan, J. Li, R. Fu, Z. Y. Lu and W. S. Yang, J. Nanosci. Nanotechnol., 2009, 9, 5874–5879 CrossRef CAS PubMed.
- Y. L. Huang, X. H. Hou, X. Y. Fan, S. M. Ma, S. J. Hu and K. Lam, Electrochim. Acta, 2015, 182, 1175–1187 CrossRef CAS.
- X. H. Hou, X. L. Zou, Y. L. Huang, S. J. Hu, Q. Ru and Y. M. Gao, RSC Adv., 2014, 4, 29534 RSC.
- S. M. Li, B. Wang, J. H. Liu and M. Yu, Electrochim. Acta, 2014, 129, 33–39 CrossRef CAS.
- N. Romcevic, R. Kostic, B. Haszic, M. Romcevic, I. K. Kudelska, W. D. Dobrowolski, U. Narkiewicz and D. Sibera, J. Alloys Compd., 2010, 507, 386–390 CrossRef CAS.
- R. Y. Zhang, X. Yang, D. Zhang, H. L. Qiu, Q. Fu, H. Na, Z. D. Guo, F. Du, G. Chen and Y. J. Wei, J. Power Sources, 2015, 285, 227–234 CrossRef CAS.
- L. Lian, L. R. Hou, L. Zhou, L. S. Wang and C. Z. Yuan, RSC Adv., 2014, 4, 49212 RSC.
- Y. D. Mo, Q. Ru, J. F. Chen, X. Song, L. Y. Guo, S. J. Hu and S. M. Peng, J. Mater. Chem. A, 2015, 3, 19765–19773 CAS.
- L. M. Yao, X. H. Hou, S. J. Hu, Q. Ru, X. Q. Tang, L. Z. Zhao and D. W. Sun, J. Solid State Electrochem., 2013, 17, 2055–2060 CrossRef CAS.
- H. Y. Yue, Q. X. Wang, Z. P. Shi, C. Ma, Y. M. Ding, N. N. Huo, J. Zhang and S. T. Yang, Electrochim. Acta, 2015, 180, 622–628 CrossRef CAS.
- C. Chen, Q. Ru, S. J. Hu, B. N. An, X. Song and X. H. Hou, Electrochim. Acta, 2015, 151, 203–213 CrossRef CAS.
- N. Yan, L. Hu, Y. Li, Y. Wang, H. Zhong, X. Hu, X. Kong and Q. Chen, J. Phys. Chem. C, 2012, 116, 7227–7235 CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.