
Phosphatidic acid-functionalized monolithic stationary phase for reversed-phase/cation-exchange mixed mode chromatography†
Kun Peng‡
abc,
Qiqin Wang‡c,
Weijia Chenc,
Donghai Xiac,
Zhengyin Zhouc,
Yuqiang Wangc,
Zhengjin Jiang*c and
Fuhai Wu*b
aSchool of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, PR China
bSchool of Public Health, Guangdong Pharmaceutical University, Guangzhou 510006, PR China. E-mail: fuhaiwu@163.com
cInstitute of Pharmaceutical Analysis, College of Pharmacy, Jinan University, Guangzhou, Guangdong 510632, China. E-mail: jzjjackson@hotmail.com
First published on 17th October 2016
Abstract
A novel phosphatidic acid functionalized polymeric monolithic column was prepared through the thermally initiated co-polymerization of 12-methacryloyl dodecylphosphatidic acid (MDPA) and ethylene glycol dimethacrylate (EDMA) in the presence of 1,4-butanediol and isopropanol as porogens within 100 μm I.D. capillaries. The polymerization conditions of monolithic columns were systematically optimized in order to obtain good permeability, stability and column efficiency. The reproducibility of the optimized monolithic column was also satisfactory. The physicochemical properties of the monolithic column were evaluated by use of instrumental techniques including scanning electron microscopy, Fourier transform infrared spectra, ζ-potential analysis and micro-HPLC. A series of test compounds such as small peptides, alkylphenones, etc., were employed to investigate the retention mechanism on the poly(MDPA-co-EDMA) monolithic column. The results demonstrate that both hydrophobic and cation-exchange interactions could contribute to the overall retention of analytes. Furthermore, the novel reversed-phase/cation-exchange mixed mode monolithic column was applied to the separations of small peptides, phenols, water-soluble vitamins B, and pharmaceutical compounds. The successful applications indicate the potential of the poly(MDPA-co-EDMA) monolithic column in complex sample analysis.
Introduction
Recently, phospholipid functionalized stationary phases have received great attention and become an important tool for pharmaceutical industry and academia with numerous applications, such as predicting the drug-cell membrane interactions, purification of membrane protein and separation of small molecules.1–3 So far, only phosphatidylcholine (PC) functionalized silica columns are commercially available. A few attempts have been directed to develop other phospholipids functionalized stationary phases.1,4,5 Phosphatidic acid (PA), the simplest anionic diacylglycerophospholipid, possesses long alkyl chains and phosphate groups and exhibits versatile complexation abilities, which is essential for the proper functioning of several transmembrane proteins and the physiology of cell membrane.2,6 Davies-Tuck et al.2 developed a strategy to immobilize PA onto 3-isothiocyanatopropyltriethoxy modified ZOR-BAX Rx-SIL silica particles for the separation of membrane peptides and proteins with different size, charge and hydrophobicity. The retention of membrane peptides and proteins on this PA functionalized column was governed by both hydrophobic and cation-exchange interactions. More recently, Bocian et al.7 prepared a series of alkyl (C10 or C18)-phosphate functionalized silica columns, which have an opposite spatial arrangement of long alkyl chains and phosphate groups. Good peak shapes and highly selectivity for β-blockers and nucleosides were obtained on these columns. It was found that the hydrophobic interaction is responsible for the retention and separation of these analytes. Therefore, developing PA functionalized stationary phases could not only enrich the type of phospholipids functionalized column for better mimicking the drug-cell membrane interactions, but also provide unique reversed-phase/cation-exchange mixed mode columns for the specific and selective separation of a wide range of compounds. The reversed-phase/cation-exchange mix mode technique has been growing rapidly because it offers more than one retention modes to improve the selectivity for simultaneous separation of complicated samples.8,9 So far, most reported PA functionalized stationary phases are silica based columns. The residual silanol and/or amino groups on these silica based stationary phases may influence their stability and the chromatographic behavior of small molecules.10,11 End-capping with glycidol/methylglycolate (MG) or decanoic/propionic anhydride could be employed to overcome this issue.1,2,7 However, the preparation process is rather complicated, which could limit its further applications involved self-lab preparation or chemical modification of silica-based phospholipid stationary phases.1 It is indeed meaningful to develop new strategies for preparing PA functionalized stationary phases.In the last decade, monolithic columns have proved to be an effective alternative to packed columns and have attracted considerable interest, owing to their facile preparation methodology, good column characteristics with respect to permeability, efficiency, stability and selectivity.12–18 Few attempts have been made to develop phospholipids functionalized monolithic columns. Jiang et al.3,19 prepared single chain PC and mixed phospholipids (single PC and phosphatidylserine (PS)) functionalized monolithic stationary phases for predicting drug-cell membrane interactions and screening the phospholipidosis-inducing potential of pharmaceuticals, respectively. However, few studies have been focused on PA functionalized monolithic columns. Zou et al.20 prepared an ethylene glycol methacrylate phosphate (EGMP) functionalized monolithic column and then coupled it with a reversed phase column for online multidimensional separation of the tryptic digest of yeast protein. Moreover, EGMP functionalized monolithic column was also extensively applied to develop immobilized metal ion affinity chromatography for phosphopeptides enrichment.21 These results demonstrated the possibility and prospect to prepare PA functionalized monolithic columns with long alkyl chains.
In this study, a phosphatidic acid monomer 12-methacryloyl dodecylphosphatidic acid (MDPA) was synthesized and employed to prepare a poly(MDPA-co-ethylene glycol dimethacrylate), (poly(MDPA-co-EDMA)) monolithic column (Fig. 1). The composition of the polymerization mixture was carefully optimized in order to obtain satisfactory column permeability, efficiency and selectivity. The retention mechanism of the poly(MDPA-co-EDMA) monolithic column was also systemically investigated using a series of small peptides as probe. Finally, the applicability of this monolithic column was evaluated using a variety of analytes.
 | ||
| Fig. 1 Synthesis of monomer MDPA and preparation of the poly(MDPA-co-EDMA) monolithic columns. Reagents and conditions: (i) methacryloyl chloride, pyridine (rt); (ii) POCl3 (0 °C); (iii) H2O, (from 0 °C to rt). | ||
Experimental
Materials and chemicals
EDMA, 2,2′-azobis(2-methylpropionitrile) (AIBN), 3-(trimethoxysilyl)propyl methacrylate (γ-MAPS), 1,4-butanediol, isopropanol, ammonium formate, formic acid, methacryloyl chloride, 1,12-dodecanediol, trimethylamine, and some test analysts (including toluene, thiourea, dimethylphthalate, anisole, naphthalene, acetophenone, propiophenone, butyrophenone, valerophenone, hexanophenone, heptanophenone, octanophenone, vitamin B12 (VB12), paracetamol, phenol, p-nitrophenol, p-bromophenol, acetylsalicylic acid, caffeine, sulfanilamide, benzenesulfonamide, phenacetin, coumarin, ranitidine, and sulpiride) were purchased from Aladdin Chemicals (Shanghai, China). Ala-Gly-Gly, Leu-Gly-Gly, Gly-Leu and Gly-Phe were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan), while Ala-Val and Leu-Val were purchased from Bachem AG (Bubendorf, Switzerland). Vitamin B1 (VB1), vitamin B6 (VB6) and vitamin B4 (VB4) were obtained from Energy chemical (Shanghai, China). The chemical structures of some test compounds were presented in Fig. S1.† Phosphorus oxychloride was purchased from Aike Reagent Corp. (Chengdu, China). Cytochrome C (Cyt-C) was obtained from Sigma-Aldrich (St. Louis, MO, USA) and the tryptic digest procedure of Cyt-C was according to previously reported method.20 HPLC-grade methanol (MeOH) and acetonitrile (ACN) were supplied by Merck (Shanghai, China). The distilled H2O used throughout all experiments was deionized and purified using a Milli-Q system (Bedford, MA, USA) and filtered through a 0.22 μm membrane before use. The fused silica capillaries with a dimension of 100 μm I.D. (375 μm O.D.) were purchased from Yongnian Optic Fiber Plant (Hebei, China).Instrumentation
1H NMR spectra was carried out using tetramethylsilane (TMS) as the internal standard in CDCl3 with a Bruker-AVANCE III Digital NMR Spectrometer (Karlsruhe, Germany) at 300 MHz. Mass spectra (MS) was performed on a Agilent 1260 Infinity Hybrid SFC/UHPLC analytical system (Santa Clara, CA, USA) coupled with a Agilent 6130 single quadrupole mass spectrometry detector (Santa Clara, CA, USA). A Jinghong DK-S22 water bath (Shanghai, China) was used for thermally initiated copolymerization. A Leo 1530 VP Field Emission Scanning Electron Microscope, equipped with Oxford INCA 400 energy dispersive X-ray microanalysis (Wiesbaden, Germany), was employed to evaluate the morphology of the monolithic stationary phases. The ζ-potential values of monolithic materials were measured by Nano-ZS ζ-potential meter (Malvern, UK). Fourier transform infrared (FT-IR) spectra were obtained on a JASCO FT/IR-4600 spectrophotometer (Tokyo, Japan) within the wavenumber range of 4000–400 cm−1 using KBr pellets. All micro-HPLC experiments were performed on a Dionex Ultimate 3000 RSLC nano system (Sunnyvale, CA, USA) consisted of an Ultimate 3000 RS variable wavelength detector with a 3 nL flow cell, an Ultimate 3000 Binary Rapid Separation nano flow pump, an Ultimate 3000 RS autosampler and a four-port injection valve with 20 nL internal loop (Houston, TX, USA). Data acquisition and date handling were carried out using Chromeleon 6.8. All chromatograms were converted into .txt files and then redrawn using Microcal Origin 8.5. The pH values were monitored using a Sartorius PB-10 pH meter (Gottingen, Germany).Synthesis of the monomer MDPA
MDPA was synthesized according to a previously reported method with proper modification (Fig. 1).22 Firstly, the intermediate 1 (3.0 g, 10 mmol) was synthesized as described previously,22 and then dissolved in 20 mL of phosphorus oxychloride. The reaction mixture was stirred at room temperature for 12 h, and it was then poured into 200 mL ice water until a large amount of white precipitates formed. The solution was directly extracted with ethyl acetate thrice without filtration, and the organic layer was collected and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (CH2Cl2/MeOH = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give the compound 2 as a straw yellow oil (2.1 g, 60% yield). ESI-MS m/z: 350.9 [M + H]−. 1H NMR (300 MHz, CDCl3) δ 6.11 (s, 1H), 5.56 (s, 1H), 4.14 (t, 2H), 3.78 (t, 2H), 1.96 (s, 3H), 1.75–1.60 (m, 4H), 1.43–1.18 (m, 16H).
:1) to give the compound 2 as a straw yellow oil (2.1 g, 60% yield). ESI-MS m/z: 350.9 [M + H]−. 1H NMR (300 MHz, CDCl3) δ 6.11 (s, 1H), 5.56 (s, 1H), 4.14 (t, 2H), 3.78 (t, 2H), 1.96 (s, 3H), 1.75–1.60 (m, 4H), 1.43–1.18 (m, 16H).
Preparation of the poly(MDPA-co-EDMA) monolithic column
Prior to the copolymerization, the inner wall of the fused-silica capillaries (100 μm I.D.) were pretreated with γ-MAPS according to the previously described method.23 The polymerization mixtures used for the preparation of the poly(MDPA-co-EDMA) monolithic column was consisted of the functional monomer (MDPA), the crosslinker (EDMA), the initiator (AIBN, ∼1 wt% with respect to the monomers) and the porogens (1,4-butanediol and isopropanol). Before introducing it into the capillary, the polymerization mixture was degassed by sonicating for 15 min. The sonication also facilitate the solubility of the monomers. The homogeneous polymerization mixture was then manually introduced into the pretreated capillary. Both ends of the capillary were sealed with GC septa. The filled capillary and the sealed 2 mL vial containing the rest of polymerization mixture were submerged into a water bath at 60 °C for 12 h. The copolymerized monolithic column was then rinsed with MeOH to flush out the porogens and residual reagents.A 1 cm length of monolithic column was cut for scanning electron microscopy (SEM) analysis. The bulk polymer in the 2 mL vial was cut into small pieces, Soxhlet extracted with MeOH for 16 h, and then dried under vacuum at 60 °C. These polymers were used for ζ-potential measurements and FI-IR spectra.
Chromatographic conditions
The mobile phases were prepared by mixing appropriate proportions (v/v) of buffer solution, H2O and ACN. The stock buffer solutions of ammonium formate were prepared by dissolving an appropriate amount of the salt in deionized H2O and adjusted to desired pH with formic acid. Unless otherwise stated, the mobile phase pH values mentioned in this research refer to the aqueous portion only. The stock sample solutions were prepared by dissolving appropriate amount of samples into MeOH to give a concentration around 1 mg mL−1. Both mobile phases and sample solutions were subjected to a filtration through 0.22 μm membrane filter prior to the HPLC experiments. Chromatograms were recorded at wavelength of 214 or 254 nm.Calculation
Permeability (K, m2) of the monolithic column was calculated according to Darcy's law by using the following eqn (1):24
 | (1) |
Chemical stability of the monolithic column can be evaluated through the swelling propensity (SP) test. The SP factor was calculated using eqn (2):25
 | (2) |
Results and discussion
Preparation of the poly(MDPA-co-EDMA) monolithic column
Because the composition of the polymerization mixture significantly affects the permeability, morphology, efficiency and selectivity of the monolithic column, several factors including the composition and the weight content of porogens, and the weight content of the crosslinker EDMA were systemically optimized by evaluating the physicochemical properties and morphology of the resulting monolithic columns using micro-HPLC (Table S1†) and SEM (Fig. 2). | ||
| Fig. 2 Scanning electron microphotographs of the poly(MDPA-co-EDMA) monolithic column. | ||
The effect of the porogens composition was first investigated by varying the weight ratio of isopropanol and 1,4-butanediol (w/w) from 86/14 (column C1) to 80/20 (column C3). It was noted that the minor change of the porogens composition has a significant influence on the column backpressure. As can be seen from Table S1,† the backpressure significantly increased from 2.3 to 8.0 MPa with decreasing the weight content of isopropanol in the porogens mixture from 86% to 80%, and column C2 exhibited the highest column efficiency (20000 plates per m). Therefore the weight ratio of 83/17 between isopropanol and 1,4-butanediol was selected for further experiments. It was also observed that the crosslinker EDMA content in the monomer mixture has some effects on the monolithic column properties. When the weight content of EDMA increased from 40% (column C4) to 50% (column C5), the column backpressure was found to rise from 2.4 MPa to 6.0 MPa and the column efficiency varied from 17000 to 6700 plates per m. An EDMA weight content of 45% (column C2) in the monomer mixture provides the highest column efficiency (20000 plates per m) and reasonable backpressure (3.1 MPa). The influence of the porogens content in the polymerization mixture was also evaluated. As can be seen in Table S1,† the weight content of porogens was increased from 55% (column C6) to 65% (column C7) (w/w), both the column backpressure and efficiency decreased. Finally the polymerization conditions used to produce column C2 were selected for all further studies.
Characterization of the poly(MDPA-co-EDMA) monolithic
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–O-alkyl stretching of monomer MDPA, respectively. These results confirmed that phosphatidic acid functionalities were successfully introduced into the monolithic matrix.
O and P–O-alkyl stretching of monomer MDPA, respectively. These results confirmed that phosphatidic acid functionalities were successfully introduced into the monolithic matrix.It is well known that the surface charge of monolithic stationary phases could play a decisive role in the retention characteristics of charged analytes.
The materials with PA functionality have a negative surface charge over a wide pH range (Fig. S3†).26 In order to further confirm the surface charge of the poly(MDPA-co-EDMA) monolithic column, which contains the phosphoric acid groups, ζ-potential measurements of the bulk polymer were performed in pH-dependent manner over a pH range of 3 to 7. As shown in (Fig. S3†), the ζ-potential values significantly decreased with increasing pH due to the amount of deprotonated phosphate group. This may also indicate the phosphatidic acid functionalities were immobilized successfully on the polymeric surface but not embodied into matrix.
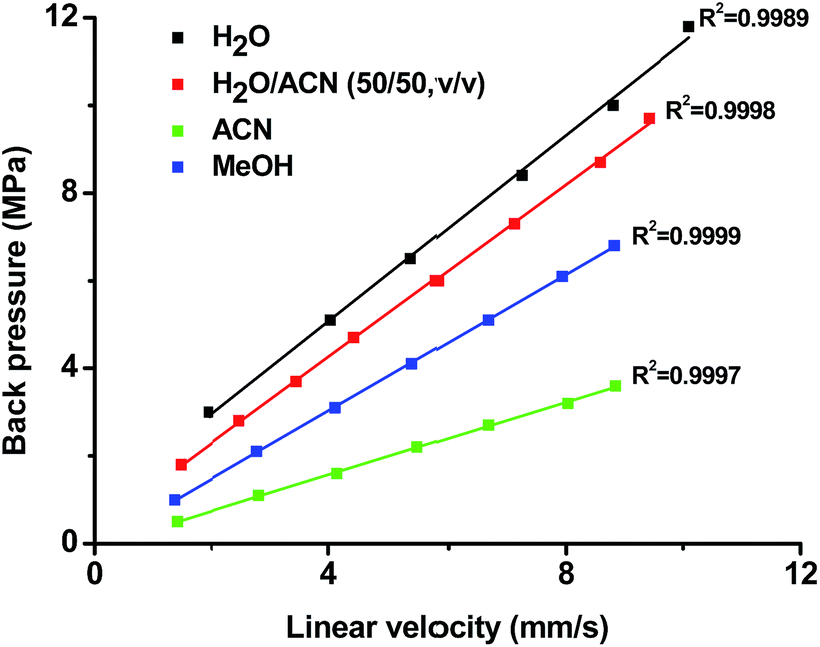 | ||
| Fig. 3 Correlation of backpressure and linear velocity for poly(MDPA-co-EDMA) monolithic column with different mobile phases. Conditions: column dimensions, 140 mm × 100 μm I.D.; mobile phase, ACN, MeOH, H2O and H2O/ACN (50/50, v/v); UV detection wavelength, 214 nm; injection volume, 20 nL; samples: toluene. | ||
The reproducibility of the optimized poly(MDPA-co-EDMA) monolithic column was finally assessed by measuring the relative standard deviations (RSDs) for the retention factors (k) of dimethyl phthalate, anisole and naphthalene (thiourea as the t0 marker) (Table S3†). A mixture of H2O/ACN (60/40, v/v) was used as the mobile phase. The RSD values for the run-to-run reproducibility (n = 10) of dimethyl phthalate, anisole and naphthalene are 0.51%, 0.34% and 0.34%, respectively, while the day-to-day reproducibility (n = 3) are 2.06%, 2.09% and 2.31%, respectively. Notably, the separation performance of the poly(MDPA-co-EDMA) monolithic column does not appear to deteriorate after 100 injections over 60 days. These results confirm the satisfactory robustness of the monolithic column. Furthermore, the column-to-column and batch-to-batch reproducibilities were also investigated with the same batch or different batches of polymerization mixture. The column-to-column reproducibility (n = 3) were 1.38%, 1.33% and 1.38% for the retention factors of dimethyl phthalate, anisole and naphthalene. The batch-to-batch reproducibility (n = 3) of three analytes were 2.82%, 2.29% and 2.11%, respectively. Moreover, RSD values for their reproducibility based on quantitative performance (peak height and peak area) were also satisfactory (Table S3†). These results further confirm the good reproducibility of the optimized poly(MDPA-co-EDMA) monolithic columns.
Separation mechanisms
 | ||
| Fig. 4 Relationship between retention time and ACN concentration on the poly(MDPA-co-EDMA) monolithic column. Conditions: column dimensions, 140 mm × 100 μm I.D.; mobile phase, H2O/ACN; flow rate, 600 nL min−1; UV detection wavelength, 214 nm; injection volume, 20 nL; samples: toluene, thiourea. | ||
The hydrophobicity of the poly(MDPA-co-EDMA) monolithic column was further evaluated through determining its methylene selectivity, which is calculated from the antilogarithm of the slope of a plot of the natural logarithm of retention factor (lnk) vs. the carbon number (n) of a set of alkylphenones.28 A mixture of H2O/ACN (50/50, v/v) was employed as the mobile phase. Under this condition, a baseline separation of all seven alkylphenones can be obtained within 14 min (Fig. 5). As depicted in Fig. S4,† a good linear correlation between lnk and n was observed and the calculated methylene selectivity is 1.53 for the poly(MDPA-co-EDMA) monolith. This further confirm the typical reversed phase retention characteristics on the poly(MDPA-co-EDMA) monolithic column.
 | ||
| Fig. 5 Chromatogram of seven alkylphenones separation. Conditions: column dimensions, 140 mm × 100 μm I.D.; mobile phase, H2O/ACN (50/50, v/v); detection wavelength, 214 nm; flow rate, 600 nL min−1; samples, (1) acetophenone; (2) propiophenone; (3) butyrophenone; (4) valerophenone; (5) hexanophenone; (6) heptanophenone; (7) octanophenone. | ||
D) of small peptides were calculated and listed in Table S4.†The effect of buffer pH on the retention of small peptides was first studied by varying the pH of ammonium formate buffer from 3.0 to 6.4, while remaining the salt concentration at 10 mM in H2O/ACN (90/10, v/v). As shown in Fig. 6a, the increase in buffer pH clearly led to a reduction in retention of six small peptides on the poly(MDPA-co-EDMA) monolithic column. Such behaviors could be explained by cation-exchange interactions. When the mobile phase pH increased from 3.0 to 6.4, the peptides became less positively charged and even negatively charged, while their hydrophobicity did not change significantly as listed in Table S4.† At the same time, the negative charge of the monolithic column increased. As a results, the electrostatic attraction interaction between peptides and the poly(MDPA-co-EDMA) monolithic stationary phase became weaker or even change to electrostatic repulsion interaction, and thus led to a shorter retention time. These results indicate that cation-exchange interaction plays a key role in the retention of small peptides on the poly(MDPA-co-EDMA) monolithic column.
 | ||
| Fig. 6 Influence of mobile phase pH, salt concentration and ACN content on retention factors (plot a–c) and chromatogram of small peptides separation (plot d). Conditions: column dimensions, 180 mm × 100 μm I.D.; flow rate, 400 nL min−1; detection wavelength, 214 nm; injection volume, 20 nL; mobile phase, plot a, H2O/ACN (90/10, v/v) containing 10 mM ammonium formate with various pH, plot b, H2O/ACN (90/10, v/v) containing ammonium formate pH 4.3 at various concentration, plot c, H2O/ACN containing 10 mM ammonium formate pH 4.3 at various ACN content, plot d, 10 mM ammonium formate, pH 4.3, in H2O/ACN (80/20, v/v); samples, (1) Ala-Gly-Gly; (2) Leu-Gly-Gly; (3) Ala-Val; (4) Gly-Leu; (5) Gly-Phe; (6) Leu-Val. | ||
The effect of ionic strength on the retention of small peptides was also investigated by varying the concentration of ammonium formate (pH 4.3) in the mobile phase of H2O/ACN (90/10, v/v). At pH 4.3, all small peptides are positively charged. As shown in Fig. 6b, the retention of small peptides decreased as the ammonium formate concentration increased from 10 to 20 mM. These behaviors could be attributed to the inhibition effect of high salt concentration in the mobile phase on the cation-exchange interaction between small peptides and the poly(MDPA-co-EDMA) monolithic stationary phase.
To further investigate its cation-exchange characteristics, a simple empirical stoichiometric displacement model was employed. The model predicts a linear dependence between k and 1/[C] given by eqn (3):29
 | (3) |
In addition, the ACN content in the mobile phase has a significant influence on the retention of small peptides. As can be seen in Fig. 6c, their retention decreased with increasing the ACN content as other conditions were kept constant, implying a typical reversed-phase retention mechanism in this case. The contribution of hydrophobic interaction to the retention could also be evidenced by their elution order, which is consistent with the order of calculated logD4.3 values.
All these results indicate that both cation-exchange and hydrophobic interactions could contribute significantly to the overall retention of charged analytes on the poly(MDPA-co-EDMA) monolithic column. This could increase the separation power and degree of freedom in adjusting the separation selectivity compared to single cation-exchange or reversed-phase mode. By optimizing the pH and ionic strength, as well as organic solvent concentration, a good separation was obtained for the six small peptides within 20 min (Fig. 6d).
Application
Phenols are important contaminants with moderate toxicity and persistence in the environment.31 The applicability of the poly(MDPA-co-EDMA) monolithic column for the separation of phenols was investigated. As shown in Fig. 7, a fast separation of four phenols was achieved within 3 min using a mixture of H2O/ACN (50/50, v/v) as the mobile phase. Paracetamol (pKa 9.38, logP 1.08) was eluted first and followed with phenol (pKa 9.86, logP 1.48), p-nitrophenol (pKa 7.13, logP 1.91), and p-bromophenol (pKa 9.34, logP 2.49) in turns. This elution order is consistent with the order of their logP values. Since the majority of phenols (apart from p-nitrophenol) are uncharged at pH 7.0, the contributions from cation-exchange interactions are negligible under this separation conditions. Therefore, the hydrophobic interaction may dominate the retention behaviour of these phenols.
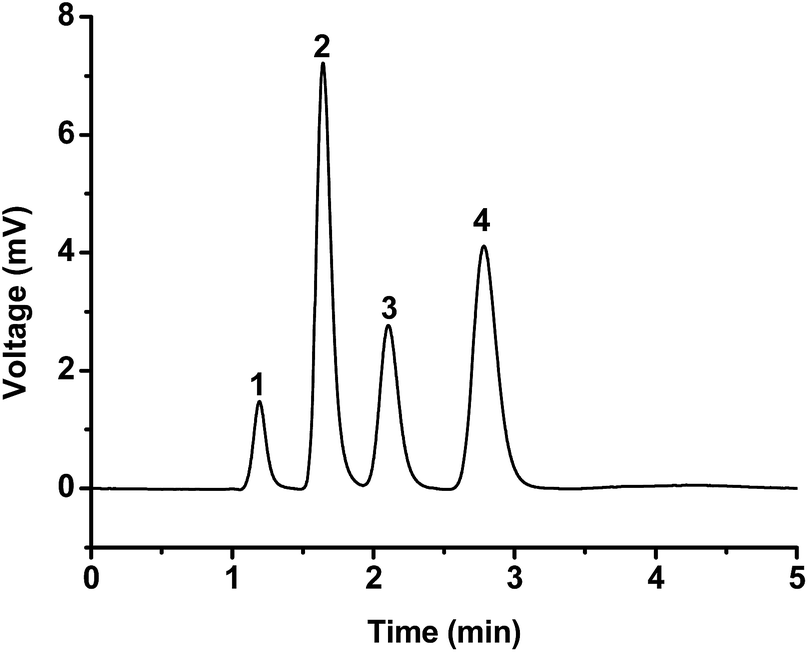 | ||
| Fig. 7 Chromatogram of phenols separation. Conditions: column dimensions, 140 mm × 100 μm I.D.; mobile phase, H2O/ACN (50/50, v/v); flow rate, 600 nL min−1; detection wavelength, 214 nm; injection volume, 20 nL; samples, (1) paracetamol, (2) phenol, (3) p-nitrophenol, (4) p-bromophenol. | ||
The group of water-soluble vitamins B are important micronutrients.32 In order to evaluate the separation power of the poly(MDPA-co-EDMA) monolithic column in the micro-LC mode for water-soluble vitamins B, four vitamins (VB1, VB4, VB6 and VB12) were employed as test analytes. As depicted in Fig. 8, a baseline separation of all vitamins B was obtained using a 10 mM ammonium formate (pH 3.0) in H2O/ACN (10/90, v/v) as mobile phase. Vitamins B were eluted in the order of VB12 (pKa 1.59, logP −0.91) < VB1 (pKa 4.80, logP 1.20) < VB6 (pKa 5.58, logP −1.00) < VB4 (pKa 9.80, logP −0.03) according to their pKa values but not logP values. Due to the high ACN content in the mobile phase, the hydrophobic interaction is unlikely to contribute to the retention of analytes. The cation-exchange interaction may play a dominate role. At pH 3.0, VB12, VB1 and VB6 will be partially deprotonated and present in negative form with different degree of charge, while VB4 will be completely protonated and fully positively charged. As a result, four water-soluble vitamins B can be separated due to the different degree of the electrostatic interaction.
 | ||
| Fig. 8 Chromatogram of water-soluble vitamins B separation. Conditions: column dimensions, 140 mm × 100 μm I.D.; mobile phase: H2O/ACN (10/90, v/v) containing 10 mM ammonium formate, pH 3.0; flow rate, 600 nL min−1; detection wavelength, 254 nm; injection volume, 20 nL; samples, (1) VB12; (2) VB1; (3) VB6; (4) VB4. | ||
In order to further evaluate the separation ability of the poly(MDPA-co-EDMA) monolithic column, a mixture of eight pharmaceutical compounds with MW ranging between 146 and 342 g mol−1 (including acidic drugs: aspirin; neutral drugs: coumarin, phenacetin and ranitidine; basic drugs: sulfanilamide, benzene sulfonamide and sulpiride), was selected as test analytes. Some parameters (pKa, logD7.4) of pharmaceutical compounds were calculated and listed in Table S5.† Ammonium formate (50 mM, pH 7.4) buffer and 100% ACN were used as mobile phase component A and B, respectively. As shown in Fig. 9, all pharmaceutical compounds were baseline separated on the poly(MDPA-co-EDMA) monolithic column using a 18 min gradient of ACN from 10% to 100% in the mobile phase. The peak shapes of all pharmaceutical compounds are acceptable for further application. However, this elution order is neither consistent with the order of their pKa values nor that of logD7.4 values. The observation could be attributed to that the comprehensive influence of reversed-phase/cation-exchange mixed interactions.
 | ||
| Fig. 9 Chromatogram of pharmaceutical compounds separation. Conditions: column dimensions, 256 mm × 100 μm I.D.; mobile phase, eluent A, 50 mM ammonium formate, pH 7.4 in H2O, eluent B, ACN; linear gradient, 0 min/10% B, 15.0 min/100% B, 18.0 min/100% B, 18.5 min/10% B, 26.0 min/10% B; detection wavelength, 214 nm; flow rate, 600 nL min−1; injection volume, 20 nL; samples, (1) acetylsalicylic acid (aspirin), (2) caffeine, (3) sulfanilamide, (4) benzene sulfonamide, (5) phenacetin, (6) coumarin, (7) ranitidine, (8) sulpiride. | ||
Finally, the separation of a tryptic digestion sample of Cyt-C was performed on the monolith to evaluate its practical utility. Fig. S6† shows more than ten resolved peaks. These results further confirm the potential applicability of the poly(MDPA-co-EDMA) monolithic column for biological samples.
Conclusions
In this study, a novel porous poly(MDPA-co-EDMA) monolithic column was prepared via a one-step copolymerization of MDPA and EDMA using isopropanol and 1,4-butanediol as porogens. The optimized monolithic column showed satisfactory properties with respect to morphology, permeability, mechanical and chemical stability, and chromatographic performance. Reversed-phase and cation-exchange mixed-mode retention mechanism was observed on the poly(MDPA-co-EDMA) monolithic column. Furthermore, the resulting monolithic column was successfully applied to the separation of a wide range of compounds, such as small peptides, phenols, water-soluble vitamins B, pharmaceutical compounds. This research not only developed a novel phospholipid functionalized monolithic column for the separation of small molecular, but also provided an opportunity for mimicking the cell membrane environment containing PA.Author contributions
K. P. and Q. Q. W. contributed equally to this work. Q. Q. W., F. H. W. and Z. J. J. supervised the project and designed the experiments. K. P., Q. Q. W., W. J. C., D. H. X. and Z. Y. Z. performed experiments and analysed data. Z. J. J., Y. Q. W. and F. H. W. contributed to modification of the manuscript. K. P. and Q. Q. W. wrote the manuscript. All authors participated in discussion of scientific ideas.Acknowledgements
We gratefully appreciate the financial support from the National Natural Science Foundation of China (Grants: 81503030), the Natural Science Foundation of Guangdong Province, China (Grant No. 2015A030310175).Notes and references
- F. Tsopelas, T. Vallianatou and A. Tsantili-Kakoulidou, Expert Opin. Drug Discovery, 2016, 11, 473–488 CrossRef CAS PubMed.
- M. Davies-Tuck, T. Lee, A. Apffel and M. I. Aguilar, J. Chromatogr. A, 2007, 1156, 167–173 CrossRef CAS PubMed.
- X. Zhao, W. Chen, Z. Zhou, Q. Wang, Z. Liu, R. Moaddel and Z. Jiang, J. Chromatogr. A, 2015, 1407, 176–183 CrossRef CAS PubMed.
- C. Pidgeon, S. Ong, H. Choi and H. Liu, Anal. Chem., 1994, 66, 2701–2709 CrossRef CAS PubMed.
- S. Ong, S. J. Cai, C. Bernal, D. Rhee, X. Qiu and C. Pidgeon, Anal. Chem., 1994, 66, 782–792 CrossRef CAS PubMed.
- E. E. Kooijman and K. N. Burger, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2009, 1791, 881–888 CrossRef CAS PubMed.
- S. Bocian, A. Nowaczyk and B. Buszewski, Anal. Bioanal. Chem., 2012, 404, 731–740 CrossRef CAS PubMed.
- K. Hu, W. Zhang, H. Yang, Y. Cui, J. Zhang, W. Zhao, A. Yu and S. Zhang, Talanta, 2016, 152, 392–400 CrossRef CAS PubMed.
- Z. Jiang, N. W. Smith, P. D. Ferguson and M. R. Taylor, J. Sep. Sci., 2008, 31, 2774–2783 CrossRef CAS PubMed.
- A. Taillardat-Bertschinger, A. Galland, P. A. Carrupt and B. Testa, J. Chromatogr. A, 2002, 953, 39–53 CrossRef CAS.
- M. Amato, F. Barbato, P. Morrica, F. Quaglia and M. I. La Rotonda, Helv. Chim. Acta, 2000, 83, 2836–2847 CrossRef CAS.
- Q. Wang, J. Feng, H. Han, P. Zhu, H. Wu, M. L. Marina, J. Crommen and Z. Jiang, J. Chromatogr. A, 2014, 1363, 207–215 CrossRef CAS PubMed.
- Q. Wang, P. Zhu, M. Ruan, H. Wu, K. Peng, H. Han, G. W. Somsen, J. Crommen and Z. Jiang, J. Chromatogr. A, 2016, 1444, 64–73 CrossRef CAS PubMed.
- N. Li, Y. Shen, L. Qi, Z. Li, J. Qiao and Y. Chen, RSC Adv., 2015, 5, 61436–61439 RSC.
- H. Wu, Q. Wang, M. Ruan, K. Peng, P. Zhu, J. Crommen, H. Han and Z. Jiang, J. Pharm. Biomed. Anal., 2016, 121, 244–252 CrossRef CAS PubMed.
- Q. Wang, E. Sanchez-Lopez, H. Han, H. Wu, P. Zhu, J. Crommen, M. L. Marina and Z. Jiang, J. Chromatogr. A, 2016, 1428, 176–184 CrossRef CAS PubMed.
- R. D. Arrua and E. F. Hilder, RSC Adv., 2015, 5, 71131–71138 RSC.
- R. Poupart, D. N. E. Houda, D. Chellapermal, M. Guerrouache, B. Carbonnier and B. L. Droumaguet, RSC Adv., 2016, 6, 13614–13617 RSC.
- X. Zhao, W. Chen, Z. Liu, J. Guo, Z. Zhou, J. Crommen, R. Moaddel and Z. Jiang, J. Chromatogr. A, 2014, 1367, 99–108 CrossRef CAS PubMed.
- F. Wang, J. Dong, X. Jiang, M. Ye and H. Zou, Anal. Chem., 2007, 79, 6599–6606 CrossRef CAS PubMed.
- H. Zhou, M. Ye, J. Dong, E. Corradini, A. Cristobal, A. J. Heck, H. Zou and S. Mohammed, Nat. Protoc., 2013, 8, 461–480 CrossRef CAS PubMed.
- J. Seuring, P. Reiss, U. Koert and S. Agarwal, Chem. Phys. Lipids, 2010, 163, 367–372 CrossRef CAS PubMed.
- N. Li, Y. Shen, L. Qi, Z. Li, J. Qiao and Y. Chen, RSC Adv., 2015, 5, 61436–61439 RSC.
- R. Yu, W. Hu, G. Lin, Q. Xiao, J. Zheng and Z. Lin, RSC Adv., 2015, 5, 9828–9836 RSC.
- F. Nevejans and M. Verzele, J. Chromatogr. A, 1985, 350, 145–150 CrossRef CAS.
- D. Marsh, CRC handbook of lipid bilayers, CRC Press, Boca Raton, Florida, 1990 Search PubMed.
- X. Lin, W. Jia, S. Feng, J. Lin and Z. Xie, RSC Adv., 2013, 3, 21888–21895 RSC.
- Q. Duan, C. Liu, Z. Liu, Z. Zhou, W. Chen, Q. Wang, J. Crommen and Z. Jiang, J. Chromatogr. A, 2014, 1345, 174–181 CrossRef CAS PubMed.
- J. Ståhlberg, J. Chromatogr. A, 1999, 855, 3–55 CrossRef.
- Z. Zhang, J. Xu, D. Hussain and Y. Q. Feng, J. Chromatogr. A, 2016, 1453, 71–77 CrossRef CAS PubMed.
- M. Cledera-Castro, A. Santos-Montes and R. Izquierdo-Hornillos, J. Chromatogr. A, 2005, 1087, 57–63 CrossRef CAS PubMed.
- H. Panahi, H. Kalal, A. Rahimi and E. Moniri, Pharm. Chem. J., 2011, 45, 125–129 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21504a |
| ‡ K. Peng and Q. Q. Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |