
Photo-Fenton process in a Co(ii)-adsorbed micellar soft-template on an alumina support for rapid methylene blue degradation†
Abstract
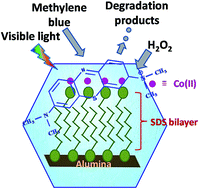
Fenton and Fenton-like processes are versatile and widely applied for organic pollutant degradation. In the present work, a new approach has been made for such a process, where ‘admicellar support’ on alumina has been used to hold the catalyst (in this case Co(II)), as well as the target pollutant methylene blue (MB). In this case, while, both are firmly attached to the layer, the active species, ˙OH radicals, generated from the reaction of Co(II) and H2O2 reacts with MB leading to degradation products in an efficient way. Visible light facilitates the reaction. Effects of various operational parameters such as dose of the catalyst, initial concentration of the dye, H2O2 concentration and light intensity, have been studied. The kinetics followed ‘zero order’ indicating a true ‘surface catalyzed reaction’. Leaching of Co(II) is minimal which is beneficial for real water treatment. The new features of this study are that the degradation of MB is entirely on a solid surface, and monitoring the reaction is done through an extraction method. The degradation products are analyzed by ESI-MS. The turn over number (TON) and turn over frequency (TOF) of the catalyst are found to be 6.82 × 1020 molecules per g and 1.89 × 1017 molecules per g per second, respectively. The recycling ability of the material is also very good up to the 3rd cycle.
 Please wait while we load your content...
Please wait while we load your content...

