
Aluminum derivative peroxides in the (t-BuO)3Al–2t-BuOOH catalytic system as a source of electron-excited dioxygen: a quantum chemical study on a model†
Oleg B. Gadzhiev*ab,
Victor A. Dodonov*a and
Alexander I. Petrovcd
aDepartment of Chemistry, N.I. Lobachevsky State University of Nizhny Novgorod, 23 Gagarin Avenue, Nizhny Novgorod, 603950, Russia. E-mail: euriscomail@mail.ru; vadodonov@gmail.com
bG.G. Devyatykh Institute of Chemistry of High-Purity Substances, Russian Academy of Sciences, 49 Troponina Street, Nizhny Novgorod, 603950, Russia
cInstitute of Non-Ferrous Metals and Materials Science, Siberian Federal University, 81 Svobodny Prospect, Krasnoyarsk, 660041, Russia
dInstitute of Chemistry and Chemical Technology, Siberian Branch of Russian Academy of Sciences, 42 K. Marx Street, Krasnoyarsk, 660049, Russia
First published on 29th September 2016
Abstract
The quantum chemical study of the MeOOH/(MeO)3Al model system has been carried out in order to predict the mechanism of the catalytic decomposition of t-BuOOH under mild conditions for the t-BuOOH/(t-BuO)3Al system being a powerful synthetic tool for selective oxidation. To elucidate the chemical excitation of O2 eliminated in the catalytic reaction and to predict the electronic state of O2, the topology of the potential energy surface (PES), the structures of intermediates and transition states, the activation and reaction energies were obtained at the B3LYP/cc-pVTZ theory level. It was shown that the peroxide, (MeO)2AlOOMe, corresponding to the experimentally obtained (t-BuO)2AlOOBu-t, is formed in the first step of the reaction. After that, in the main pathway, the aluminum-containing peroxide reacts with the second MeOOH molecule through the nucleophilic substitution of the second methoxy group forming the MeOAl(OOMe)2 diperoxide. The diperoxide rearranges to aluminum-containing ozonide MeOAlOOOMe. The ozonide isomerizes in the mononuclear-metal dioxygen intermediate (MeO)3Al·O2. The latter decomposes through the adiabatic ((MeO)3Al + O2(b1Σ+g)) and non-adiabatic ((MeO)3Al + O2(X3Σ−g)) pathways, which corresponds to experimental data about the incomplete conversion of O2 to O2(b1Σ+g). The generation of O2(b1Σ+g) was revealed by the analysis of the energy diagram calculated with the CCSD(T), CCSDT(Q), and CASSCF methods. It was suggested that the η1-(MeO)3Al·O2 and, thus, (t-BuO)3Al·O2 complexes are new sources of O2(b1Σ+g).
1. Introduction
Aluminum-containing peroxides have become of special interest for industrial organic synthesis, e.g., the original K. Ziegler's Alfol process and its modification called Epal.1 It is known that the oxidation of organoaluminum compounds (OAC) by dioxygen to the alkoxides of higher alcohols passes through the intermediate steps of forming organoaluminum peroxides: R2AlOOR, R(RO)AlOOR, and (RO)2AlOOR. The organoaluminum peroxides can be an impurity in OACs used as the essential co-compounds of catalysts for the coordination polymerization of α-olefins and other monomers. However, details of the stepwise OAC oxidation mechanism are still unknown.2,3 Chemiluminescence and, thus, chemical excitation are observed in the OAC oxidation.4 The high reactivity and low thermal stability of aluminum-containing peroxides are caused by the lability of the Al–OO fragment, which makes it difficult to isolate them individually and, thus, to study their reactivity.The investigations by V. A. Dodonov et al.5,6 of the early 1990s summarized in review7 point out that organoaluminum peroxides (RO)2AlOOR′ can be synthesized individually only with tertiary alkyl (aryl) peroxo groups (see ref. 8 and ESI†). The nucleophilic substitution reactions of aluminum tri-tert-butoxide with tertiary hydroperoxides (t-BuOOH, PhC(CH3)2OOH, Ph3COOH) have been studied further in ref. 5, 6 and 9–11 (Scheme 1). The corresponding organoaluminum peroxides have been obtained in the reactions with cumene hydroperoxide and trityl hydroperoxide in benzene at room temperature (Scheme 1).
 | ||
| Scheme 1 The high-yield synthesis of individual organoaluminum peroxides (t-BuO)2AlOOR′ from ready available (t-BuO)3Al. | ||
Unlike aryl-containing hydroperoxides, tert-butyl hydroperoxide reacts with aluminum tert-butoxide yielding dioxygen and tert-butanol. The reaction of aluminum tert-butoxide with tert-butyl hydroperoxide at the mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in benzene or CCl4 yields 80–90% of O2. The elimination of O2 remains the main reaction5,6 when the compound ratio is increased to 1:10. Thus, the catalytic reaction occurs in the (t-BuO)3Al–2t-BuOOH system, where (t-BuO)3Al is a catalyst and t-BuOOH is a reagent. The catalytic systems based on aluminum or titanium tert-butoxides and tert-butyl hydroperoxide oxidize organic sulfides to sulfones with the yield of 82–99% at room temperature and at the mole ratio of 1:2 for (t-BuO)3Al and t-BuOOH12–14 (see also ref. 15 and 16, review17 and references therein). Other examples of aluminum catalysts in a combination with H2O2 or alkyl hydroperoxide as oxidants are relatively scarce: BINOL-Al in the Bayer–Villiger reaction developed by C. Bolm et al.,18,19 Al(salelen) for sulfides (chiral)oxidation developed by T. Katsuki.20,21
:2 in benzene or CCl4 yields 80–90% of O2. The elimination of O2 remains the main reaction5,6 when the compound ratio is increased to 1:10. Thus, the catalytic reaction occurs in the (t-BuO)3Al–2t-BuOOH system, where (t-BuO)3Al is a catalyst and t-BuOOH is a reagent. The catalytic systems based on aluminum or titanium tert-butoxides and tert-butyl hydroperoxide oxidize organic sulfides to sulfones with the yield of 82–99% at room temperature and at the mole ratio of 1:2 for (t-BuO)3Al and t-BuOOH12–14 (see also ref. 15 and 16, review17 and references therein). Other examples of aluminum catalysts in a combination with H2O2 or alkyl hydroperoxide as oxidants are relatively scarce: BINOL-Al in the Bayer–Villiger reaction developed by C. Bolm et al.,18,19 Al(salelen) for sulfides (chiral)oxidation developed by T. Katsuki.20,21
In order to understand the processes in the (t-BuO)3Al–2t-BuOOH catalytic system, reactions in non-reactive solvents (benzene and carbon tetrachloride) and in oxidizable solvents (various hydrocarbons) were studied in ref. 5, 6, 9 and 11. As was shown,5,6,9,11 the dioxygen formed in the catalytic reaction decays practically completely oxidizing geminal C–H bonds in alkanes and alkylaromatic hydrocarbons. The selective oxidation of C–H bonds in alkanes and alkylarenes was studied8 by means of EPR spin trapping with 2-methyl-2-nitrosopropane (MNP) and C-phenyl-N-tert-butyl nitrone (PBN). The following oxygen-centered radicals were identified:8 t-BuO˙, t-BuOO˙, (t-BuO)2AlOO˙, and (t-BuO)2AlO˙. On the basis of the experimental results, the existence of di-tert-butoxy-tert-butyltrioxyaluminum, (t-BuO)2AlOOOBu-t, and its thermolysis mechanism were suggested (Scheme 2).6–8
 | ||
| Scheme 2 Tentative mononuclear aluminum–oxygen intermediates in the catalytic dissociation of t-BuOOH with singlet O2 generation: (t-BuO)3Al is a catalyst, t-BuOOH is a reagent. | ||
Generation of singlet dioxygen is a subject of numerous reviews,22–26 books,27,28 and ongoing researches. Indispensable is the role of singlet oxygen in photodynamic therapy of cancer, photomedicine and photobiology. Reactive oxygen substances (ROS) in ground and excited electronic states are of general importance for biochemistry and biomedical applications29–33 as well as environmental and radiochemical applications.34–36 While O2(1Δg) photosynthesis and O2 activation on transition metals' compounds are studied worldwide in many groups, the presented here catalytic chemical excitation of dioxygen up to O2(1Σ+g) on non-transition metal centre, that is, as was proposed earlier, in reactions of aluminum-containing peroxides, represents an essential novelty.
However, the proposed earlier catalytic mechanism (Scheme 2) of t-BuOOH dissociation with the active O2 generation cannot be considered as a proved mechanism because it is impossible to exclude high activation barriers for the specified reactions. It was not established earlier whether the assumed reactions are elementary ones and whether other short-lived and highly reactive intermediates containing aluminum dioxygen bond exist. Thus, the goal of the present study is to predict reaction mechanism in the (MeO)3Al + 2MeOOH system modeling the (t-BuO)3Al–2t-BuOOH system where the catalytic activation of dioxygen with the participation of a metal center was exhibited earlier. Here, we use quantum chemical methods to study structures, vibrational frequencies of intermediates and transition states and to predict reaction paths and sources of chemically excited and chemically activated O2.
2. Computational details
The topology of the potential energy surface (PES) of the (MeO)3Al + 2MeOOH system and the reaction mechanism are studied with the density functional theory (DFT) at the B3LYP/cc-pVTZ theory level.37–41 The switch to the (MeO)3Al + 2MeOOH model system obtained by substitution of the bulky tert-butyl groups by methyl groups makes it possible to study thoroughly various hypothetical reaction pathways without a considerable increase of computational time. Taking into account a conformational non-rigidity of molecules, the (MeO)3Al conformation isomer characterized by the C3h point symmetry group and two MeOOH (methylhydroperoxide) molecules were selected as the initial reagents.To investigate the electronic structure of some intermediates that are primarily important for the generation of 1O2 and the determination of the electronic state of 1O2, state-of-the art quantum chemical methods were employed. On the model system, it becomes possible to use the coupled cluster methods (up to CCSDT(Q)) and the complete active space method (CASSCF).
| 2MeOOH → 2MeOH + 3O2 | (1) |
| 2MeOOH → 2MeOH + O2(1Δg) | (2) |
| 2MeOOH → 2MeOH + O2(b1Σ+g) | (2a) |
| (MeO)3Al·O2 → (MeO)3Al + O2(1Δg) | (3) |
| (MeO)3Al·O2 → (MeO)3Al + O2(b1Σ+g) | (3a) |
| (MeO)3Al·O2 → (MeO)3Al + 3O2 | (4) |
The energies of the reactions (1) and (2) were estimated at the CCSDT(Q,fc)/cc-pVTZ theory level using the focal point analysis based on the CCSD(T,fc)/cc-pVTZ and CCSDT(Q,fc)/cc-pVDZ calculations. The energy of the reaction (2a) was calculated on the basis of the estimated reaction energy (2) and the experimental energy of the O2(b1Σ+g) ← O2(a1Δg) transition.
As will be shown in the present study, the (MeO)3Al·O2 intermediate is of especial importance for O2 generation. Thus, to establish the electronic state of 1O2 eliminated in the (MeO)3Al·O2 decomposition and to reveal the possibility of chemical electron excitation, i.e., to distinguish between the reactions (3) and (3a), the PES profile corresponding to the reaction ((MeO)3Al·O2 → (MeO)3Al + 1O2) was determined while the energy of the reaction was calculated using the CCSD(T)/6-31+G(2df,p)//B3LYP/cc-pVTZ, CCSD(T)/cc-pVTZ//B3LYP/cc-pVTZ composite approaches, and the CAS(14,10)/6-311G(d) theory level. The energies of the quasi-isolated system [(MeO)3Al + 1O2] were obtained by relaxed scans at the CAS(14,10)/6-311G(d) theory level. These reaction paths correspond to the adiabatic reaction. In these calculations, the aluminum–adjacent oxygen interatomic distance in the Al–O2 moiety was elongated to 1.5 Å above the equilibrium bond length in the (MeO)3Al·O2. Moreover, to distinguish between the reactions (3) and (3a) and, thus, to reveal an electronic term of O2, the energy of the reaction (4) was calculated at the CCSD(T)/cc-pVTZ theory level while the energies of the asymptotes corresponding to the reactions (3) and (3a), i.e., the levels of products, were estimated on the basis of the CCSDT(Q)/cc-pVTZ data for the electronic states of O2 and the coupled cluster data for the reaction (4). This procedure was applied since the calculations of (MeO)3Al·O2 with the CCSDT(Q) method are prohibitive time- and resource demanding.
To take into account the possibility of chemically activated O2 generation (reaction (4)) and, thus, to predict non-adiabatic reaction pathways in the MeOOH decomposition of the MeOOH/(MeO)3Al catalytic system, intersections between PESs were searched at the B3LYP/cc-pVTZ theory level.
The calculations were conducted with the Gaussian 03 software42 (for the B3LYP and the CASSCF methods), the GAMESS43–47 and CFOUR programs48 (for the CCSD(T) method). For the CCSDT(Q,fc)/cc-pVTZ energy estimations, the focal point analysis was carried out on the results obtained with the CFOUR and MRCC49–51 suites of programs. Minimum energy crossing point (MECP) search was conducted with ORCA suite of programs.52–54 The Moltran,55 GaussView03,56 and ChemCraft57 molecular editors were used to analyze the results.
3. Results
3.1. Reaction in the (MeO)3Al + MeOOH system
The association (MeO)3Al + MeOOH → (MeO)3Al⋯HOOMe occurs barrierlessly forming quasi-cyclic isomeric complexes 1 and 1a (Fig. 1 and 2) stabilized by the hydrogen bond (the distances between the O and H atoms are about 2.131 Å and 1.702 Å for 1 and 1a, respectively) and by the Al–O coordination bond (the distances between the Al and O atoms are about 2.031 Å and 1.991 Å for 1 and 1a, respectively). The corresponding reaction energies are −93.8 kJ mol−1 and −83.4 kJ mol−1. 1a isomerizes in the structure 1 with the activation energy of 63.5 kJ mol−1. Complex 1 is rearranged via saddle point TS1 to structure 2 that is the complex of (MeO)2AlOOMe and MeOH stabilized by the hydrogen and coordination bonds. For 1a, similar reaction leading to the monomethylperoxide was not determined despite of special efforts. The activation (Ea) and reaction (Er) energies for 1 → 2 are 56.9 kJ mol−1 and 31.1 kJ mol−1, respectively. The dissociation of 2 occurs with no transition state with Ea of 77.9 kJ mol−1; the energy level of isolated reactants, (MeO)2AlOOMe (3), MeOH, and MeOOH, is located by 15.2 kJ mol−1 higher than the initial reagents ((MeO)3Al + 2MeOOH). For peroxide 3, there is an isomer 3a (Fig. 1 and 2), which differs in the formation of the intramolecular donor–acceptor bond between Al and O atoms stabilizing the three-membered quasi-cycle of AlOO. The relative energy (Erel) of mono-η2-methyl peroxide 3a is −0.3 kJ mol−1. | ||
| Fig. 1 Profile of the singlet PES for the reaction 2MeOOH + (MeO)3Al → (MeO)Al(OOMe)2 + 2MeOH obtained at the B3LYP/cc-pVTZ theory level. The dotted and solid lines indicate the minimum energy pathways. The relative energies are in kJ mol−1. | ||
 | ||
| Fig. 2 Structures calculated by full geometry optimization at the B3LYP/cc-pVTZ theory level for the reaction 2MeOOH + (MeO)3Al → (MeO)Al(OOMe)2 + 2MeOH. Bond lengths are in Å. | ||
3.2. Elimination in the (MeO)2AlOOMe + MeOOH system
Complex 2 (Fig. 2) is coordinatively unsaturated, which allows of the attachment of the MeOOH molecule from the side opposite to the MeOH molecule bonded in the donor–acceptor complex. Structure 4 formed without activation with Er of −62.1 kJ mol−1 decomposes with the activation energy of 48.5 kJ mol−1 with no transition state. The released complex 5 is stabilized by the donor–acceptor and hydrogen bonds with a six-membered quasi-cycle. The isomerization of 5 in 6 (Fig. 1) occurs through transition state TS2 with the activation energy of (Ea) 133.4 kJ mol−1 and with Er of 48.8 kJ mol−1. 5 is the complex of bis-η2-methyl peroxide 7 and MeOH (Fig. 2). The cleavage of the hydrogen bond stabilizing the complex requires the energy of 27.6 kJ mol−1.3.3. Substitution of the methoxy group in the (MeO)2AlOOMe + MeOOH system
Contrary to the association with the formation of 4 (Fig. 1), the MeOOH molecule can attack from the side of MeOH bound in the complex. In this case, 4a (Fig. 2) is formed. Through the saddle point TS1b, it can rearrange to 4b with the activation energy (Ea) of 4.7 kJ mol−1. The generation of the prereaction complexes of several structural types results in the nonuniqueness of the pathway for the reaction in the (MeO)2AlOOMe + MeOOH + MeOH system (Fig. 2) with the generation of bis-η2-methylperoxide (structure 7, Fig. 2), i.e., with the cleavage of the peroxo bond in the O–OMe group and the concerted closing of AlOO rings with the two η2-methylperoxo moieties formation.The reaction of the second MeOOH molecule with 3 or 3a or their complexes with the MeOH molecule can occur similarly to the interaction of the first MeOOH molecule with (MeO)3Al, i.e., according to the nucleophilic substitution mechanism contrary to the rearrangement 5 → TS2 → 6 (Fig. 1) that includes the cleavage of the O–O bond in the MeOO group and successive elimination of MeOH. Another reaction pathway is determined by 4c which an isomer of 4. Complex 4c with the relative energy (Erel = −125.5 kJ mol−1) that is similar to Erel of 4 (Erel = −124.8 kJ mol−1) eliminates MeOH. In this no transition state reaction with the activation energy of 63.5 kJ mol−1 8 is formed that is the isomer of 5. Structure 8 (Fig. 1 and 3) with a four-membered quasi-cycle consisting of the coordination and hydrogen bonds is 14.3 kJ mol−1 less stable than 5 (Fig. 1 and 2) formed by the coordination of the MeOOH molecule which is involved in the six-membered quasi-cycle. The isomerization 8 → TS3 → 9 occurs with the activation energy (Ea) of 34.9 kJ mol−1 and is a weakly exothermic reaction (Er = −29.9 kJ mol−1). Then, during the elimination of MeOH from 9, one or two peroxo groups may close in three-membered cycle or two cycles, respectively, with simultaneous formation the hydrogen-bonded complexes similar to 6.
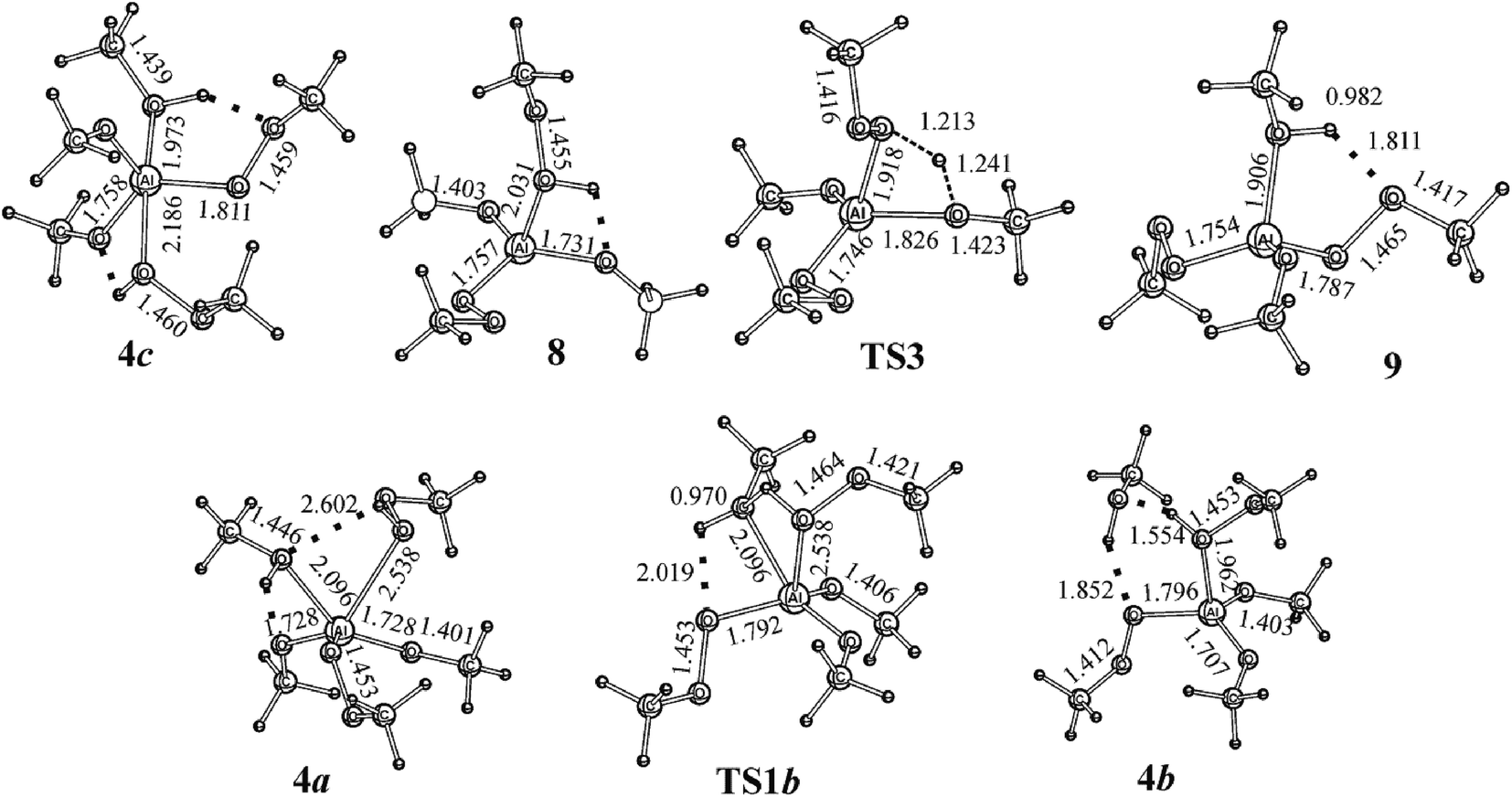 | ||
| Fig. 3 Structures calculated by full geometry optimization at the B3LYP/cc-pVTZ theory level for the reactions in the (MeO)2AlOOMe + MeOOH + MeOH system. Bond lengths are in Å. | ||
3.4. Reactions of isomeric mono- and bis-η2-methyl peroxides MeOAl(OOMe)2
Let us dwell on the reactivity of 7 (Fig. 2). The intramolecular transfer of O atom from one η2-methyl peroxo group to another η2-methyl peroxo group with the formation of ozonide-1,3 (10) is a weakly exothermic (Er = −40.5 kJ mol−1) elementary reaction with high activation energy (Ea = 182.2 kJ mol−1). The four-membered cycle in 10 (Fig. 4) is opened over the longest O–O bond (re(O–O) = 2.264 Å) via transition state TS5 with the activation energy of 37.4 kJ mol−1. The reaction 10 → 11 (Fig. 4 and 5) is practically athermic (Er = 2.9 kJ mol−1). For the fully optimized structure 11, the O atom of the η1-O2 group is not located exactly above the Al atom, but it is slightly shifted towards one of the methoxy groups, the valence angle ∠OAlO is 78° in this case. 11 is a η1-complex of the O2 and (MeO)3Al monomers, i.e., its monomolecular decay on the isolated monomers proceeds without saddle point as was exhibited by the relaxed scan at the B3LYP/cc-pVTZ theory level where smooth potential energy curve corresponding to the Al–OO bond cleavage was obtained. In other words, the association reaction O2 + (MeO)3Al → 11 is barrierless. | ||
| Fig. 4 Profile of the singlet PES corresponding to the reaction in the (MeO)Al(OOMe)2 + 2MeOH system obtained at the B3LYP/cc-pVTZ theory level. The dotted and solid lines indicate the minimum energy pathways. The relative energies are in kJ mol−1. | ||
 | ||
| Fig. 5 Structures calculated by full geometry optimization at the B3LYP/cc-pVTZ theory level for the reaction of (MeO)2AlOOMe. Bond lengths are in Å. | ||
For 7 (Fig. 2), several isomers can be obtained by opening one or two three-membered quasi-cycles AlOO (Fig. 4). Mono-η2-methyl peroxides, except for conformation isomers formed by methyl group rotation, have conformation isomers 12 and 12a (Fig. 4) which are different in the dihedral angle ∠O–Al–O–O, where O–O is a peroxo group corresponding to the opened cycle. This angle is −11.6° for 12 and −179.0° for 12a (Fig. 4). The 12 and 12a conformers are characterized by a comparable energetic stability. Indeed, the relative energy (Erel) of 12 and two MeOH monomers is 7.5 kJ mol−1 and Erel = 4.6 kJ mol−1 was calculated for 12a and two MeOH monomers (Fig. 4). The two OOAl cycles (7 → 12b) are opened with Er = 29.3 kJ mol−1 (Fig. 4). In all the cases (7.4 kJ mol−1 for 7 → 12 and 4.5 kJ mol−1 for 7 → 12a), the activation energy is many times less than Ea of the reaction 7 → TS4 → 10 (Fig. 4). If two peroxo groups in MeOAl(OOMe)2 are not closed in a cycle, the conformation isomer 12b with two MeOH monomers (Fig. 4) is 29.4 kJ mol−1 less energetically stable than the initial reagents ((MeO)3Al + 2MeOOH).
Mono-η2-methyl peroxide 12 (Erel = 7.5 kJ mol−1) is isomerized with Er = 13.5 kJ mol−1 to ozonide-1,3 (13) via transition state TS6 with the relative energy (Erel) of 129.3 kJ mol−1 (Fig. 3 and 4). Ozonide-1,3 (10) is more energetically favorable than 13. Indeed, Er calculated for the reaction 13 → 10 is 61.4 kJ mol−1; however, isomerization through TS7 (Fig. 4) with the methyl group shift is a reaction with a rather high barrier (Ea = 185.9 kJ mol−1). This activation energy value is insignificantly higher than Ea = 182.0 kJ mol−1 for the reaction pathway to 7 → 10.
12a is slightly different in reactivity from the conformer 12 (Fig. 5). In this case, the transfer of the O atom of the OO-η2 group to the peroxo group and the increase of the oxygen chain length occurs with the activation energy of 159.2 kJ mol−1 for TS6a, i.e., 12 is more reactive because Ea for 7 → 10 is 22.8 kJ mol−1 more. The OOOCH3 chain is stabilized in the form of the η2-donor–acceptor coordination complex 13a that is 41.4 kJ mol−1 more energetically stable than 13. Ozonide-1,2 (structure 13a, Fig. 4 and 5) turns out to be 20 kJ mol−1 less stable than ozonide-1,3 (10, Fig. 5). Contrary to the reaction 12 → 13, the isomerization 12a → 13a is an exothermic (Er = 25.0 kJ mol−1).
3.5. Reaction of mono-η2-methyl peroxide with MeOOH
Contrary to 5 and 8, where the coordination of the oxygen atoms of the OH and OOH groups to the Al atom and the formation of 6- and 4-membered quasi-cycles stabilize the structures, another structure can be predicted, namely, a mono-η2-methyl peroxo complex with the hydrogen bonded MeOOH. Indeed, 14 with MeOH (Fig. 6 and 7) is energetically more stable than the initial reagents (Erel = −29.3 kJ mol−1). The energy of the hydrogen bond in the complex 14 is 35.7 kJ mol−1 as calculated at the B3LYP/cc-pVTZ theory level. The isomerization 14 → 15 (Fig. 6 and 7) occurs with Ea = 173.7 kJ mol−1 through transition state TS8 (Fig. 7). Structure 15 is a complex of the MeOH molecule and (MeO)2AlOOOMe. 15 is stabilized by the hydrogen bond which occurs between the middle O atom in the AlOOOCH3 chain and HO-group of the MeOH molecule. The hydrogen bond can be formed both with the O atom of the OCH3 group and with the O atom of the OAl group. The isomerization 15 → 15a (Fig. 6 and 7) is an exothermic process (Er = −22.5 kJ mol−1); the second reaction (15 → 15b) is athermic (Er = −0.1 kJ mol−1). When MeOH is eliminated from 15 and isomeric 15a or 15b, quasi-cycles are closed with the formation of 10 and 13a.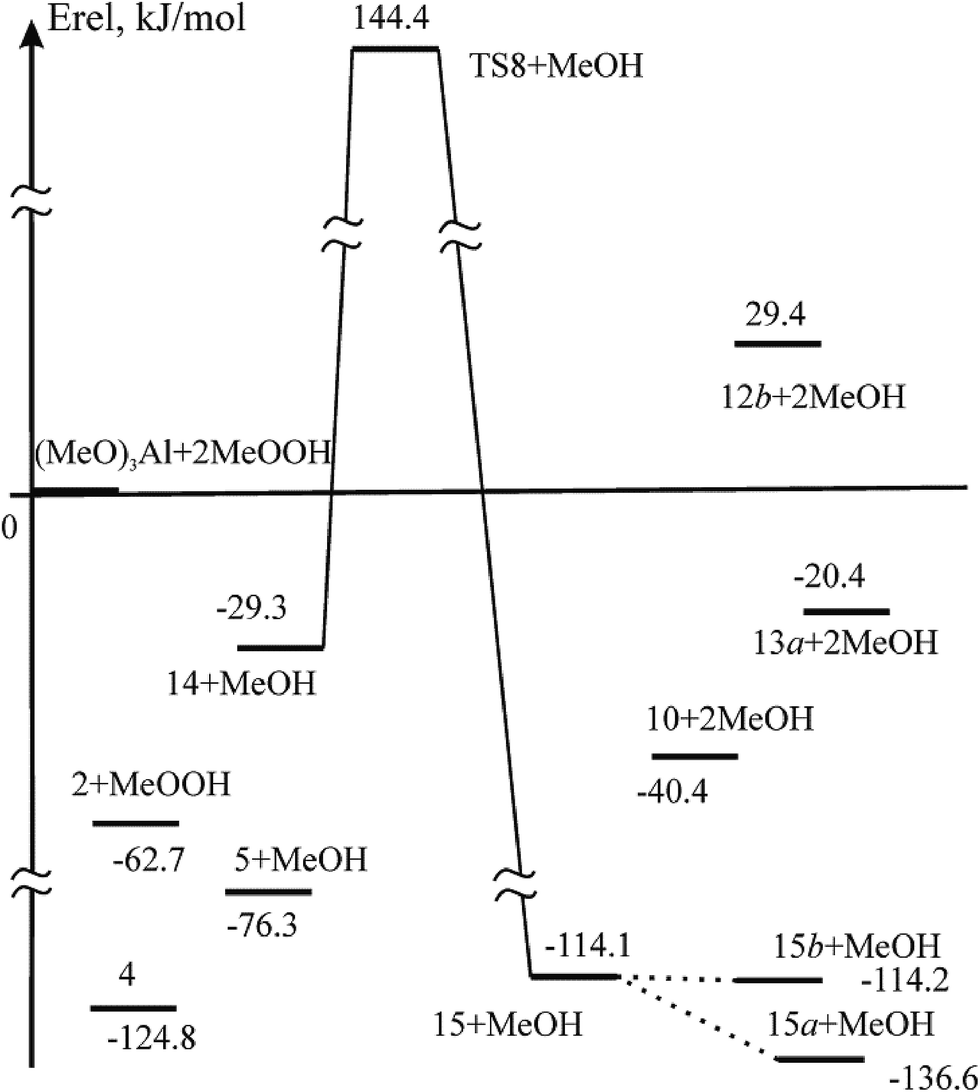 | ||
| Fig. 6 Profile of the singlet PES for the reaction in the (MeO)2AlOOMe⋯MeOOH system obtained at the B3LYP/cc-pVTZ theory level. The dotted and solid lines indicate the minimum energy pathways. The relative energies are in kJ mol−1. | ||
 | ||
| Fig. 7 Structures calculated by full geometry optimization at the B3LYP/cc-pVTZ theory level for the reaction of (MeO)2AlOOMe with MeOOH. Bond lengths are in Å. | ||
4. Discussion
4.1. Comparison of reaction pathways
The reaction pathways in the (MeO)3Al + 2MeOOH system predicted in the quantum chemical study of the PES at the B3LYP/cc-pVTZ theory level can be divided into two groups: (1) with the formation of bis-methyl peroxides (bis-η2-, mono-η2- and acyclic (MeO)2AlO2Me) that are rearranged to isomeric ozonides (MeO)2AlO3Me; (2) with the attack of MeOOH onto the AlO2Me three-membered ring of the mono-methyl peroxide, (MeO)2AlO2Me, forming the isomeric ozonides, (MeO)2AlO3Me. The ozonides are precursors for the reactive η1-O2-complex, (MeO)3Al·O2, that decomposes forming 1O2 and regenerating the catalyst, (MeO)3Al. It is worth noting that these pathways have in common the formation of isomeric ozonides or their donor–acceptor complexes with MeOH and the formation of η2-methyl peroxides ((MeO)2AlO2Me or MeOAl(O2Me)2 depicted in Fig. 1). Therefore, mono-η2- and bis-η2-methyl peroxides are formed in different stages of different reaction pathways (Fig. 1 and 6). These η2-methyl peroxides differ from acyclic isomers in energetic stability. Indeed, closing one ring in the molecule stabilizes the structure by about 15 kJ mol−1. Besides structure stabilization, closing the AlOO three-membered ring activates the O-atom of methyl peroxide. In this case, the O atom inserts in the Al–O bond of the another η2-methyl peroxo group, AlO2Me.However, the activation energy (182.0 kJ mol−1) for the intramolecular transfer of the O atom in 7 with the formation of ozonide-1,3 (10) is by 48.6 kJ mol−1 higher than the highest barrier for the net reaction 5 → 7 (Fig. 1) while the synchronous formation of two η2-groups (5 → 6, Fig. 1) is the reaction with the highest activation barrier in the PES region corresponding to the pathway of the successive substitution of two MeO in (MeO)3Al (Fig. 1). Indeed, the activation energy (Ea = 133.4 kJ mol−1) of the reaction 5 → 6 (Fig. 1) is more than two times higher than Ea = 56.9 kJ mol−1 of the reaction 1 → 2 (Fig. 1) and the total activation energy of the stepwise reaction 4c → 9 (Fig. 1).
Another reaction path to ozonide to be compared is a path with the intermediate 9. Intermediate 9 (Fig. 1) is a precursor of conformers 12 and 12a (Fig. 4) that are formed from 9 after the elimination of the coordinative bonded MeOH and the closing of one AlO2Me ring with the activation energy of 96.5 kJ mol−1 with no transition state. The reaction 12 → 13 results in the formation of the OOO chain stabilized by including in the four-membered cycle AlO3 with almost equivalent lengths of the corresponding bonds (the difference is not more than 0.0004 Å, Fig. 5). Structure 13 (Fig. 5) insignificantly differs in energy (0.1 kJ mol−1 more stable) and in geometric parameters (the value of root mean square deviation for atomic positions is 0.039 Å) from a more symmetric structure that is characterized by the Cs symmetry point group. However, the symmetric structure has one (soft) imaginary vibrational frequency which breaks the symmetry. 13 (Fig. 4) is less energetically stable than 10 (Er = −61.4 kJ mol−1 for this rearrangement), the methyl group shift requires overcoming of the barrier of 185.9 kJ mol−1 (Fig. 4). This Ea value is insignificantly higher than Ea (182.0 kJ mol−1) of the rate-determining step for the minor pathway with TS4 (Fig. 4). While the barrier of 185.9 kJ mol−1 for the two-step reaction 12 → 13 → 10 is comparable to the barrier height of 182.0 kJ mol−1 in the single-step reaction 7 → TS4 → 10 determined relative to 7, the 12 → 13 reaction is endothermic and, thus, less thermodynamically favorable. Therefore, the pathway through TS4 is more probable than the two-step reaction with TS6 and TS7.
The ozonide-1,2 (structure 13a, Fig. 4 and 5) is formed with the activation energy (Ea) of 159.2 kJ mol−1, which exceeds the activation energies in the reaction pathways of the successive (two-step) nucleophilic substitution of one methoxy group and the subsequent O–O bond cleavage closing the AlOOOMe ring. The activation barrier (7.8 kJ mol−1) of the ozonide-1,2 isomerization, (13a) → ozonide-1,3 (10), is negligible in comparison with the activation energies calculated for 12a → TS6a → 13a, the isomerization 10 → TS5 → 11, and the 1O2 elimination from the (MeO)3Al·O2 complex (11) (Fig. 3). Therefore, the pathway 12a → TS6a → 13a → TS7a → 10 → TS5 → 11 (Fig. 3) is a more probable one among the three pathways of the ozonide synthesis. Indeed, the activation energy of 159.2 kJ mol−1 for the rate-determining step (12a → TS6a → 13a), and the reaction pathway 7 → TS4 → 10 → TS5 → 11 → 1O2 + (MeO)3Al with the rate-determining step 7 → TS4 → 10 for which Ea = 182.0 kJ mol−1 is a minor channel (Fig. 4).
The reaction 14 → TS8 → 15 (Fig. 6) is a pathway to the ozonide 15 through the direct attack of MeOOH onto the mono-η2-methyl peroxo group with building the OOO chain. The activation energy (Ea) of the reaction 14 → 15 is 173.7 kJ mol−1 (Fig. 6). This value is 14.5 kJ mol−1 higher than Ea for the reaction pathway with TS6a, but it is 8.3 kJ mol−1 less than Ea of the reaction through TS4, that is, the minor pathway with TS4 is less effective than this one with TS6a (Fig. 4).
Thus, the effectively competing reaction pathways to the ozonides and their complexes were predicted at the B3LYP/cc-pVTZ theory level. In each case, it was shown that the formation of a three-oxygen-atom chain (OOO) is the rate-determining step of the net reaction in the 2MeOOH + (MeO)3Al catalytic system. It is worth noting that the reaction pathway including the isomerization of mono-η2-peroxo aluminum (12a) to ozonide-1,2 (13a) is characterized by the lowest barrier height (Ea = 159.2 kJ mol−1, Fig. 4) and, thus, it is a major adiabatic channel.
4.2. On the possibility of the chemically excited O2 formation. Whether it is O2(1Δg) or O2(b1Σ+g)?
In order to establish the electronic configuration of O2, i.e., the possibility of chemical electron excitation or chemical activation, in the decomposition of MeOOH in the MeOOH/(MeO)3Al catalytic system which initiated the sequence of elementary reactions with the participation of the mono-η2-, bis-η2-methyl peroxo aluminum compounds, the ozonides, and (MeO)3Al·O2 (η1-complex of O2), the energies of the reactions (1) and (2) were calculated with high accuracy and the singlet PES region corresponding to the decomposition (MeO)3Al·O2 → (MeO)3Al + 1O2 was scanned.As was shown in the scrupulous study of oxygen allotropes in the form of molecules and complexes,58 the energy of the transition O2(a1Δg) ← O2(X3Σ−g) is 99.5 ± 6.9 kJ mol−1 as calculated with the CCSDT(Q) method that agrees well with the experimental value59 of 94.7 kJ mol−1. In this case, it is necessary to use the CCSDT(Q) method to calculate the energy of the reaction with O2(1Δg) formation, i.e., for the reaction (2). The energy of the reaction (2a) will be obtained if we know the high accurate energy estimations of the reactions (1) and (2) and also the experimental energy of O2(b1Σ+g) ← O2(X3Σ−g).
In the present study, the energies of the reactions (1) and (2) were estimated at the CCSDT(Q,fc)/cc-pVTZ theory level using the focal point analysis based on the CCSD(T,fc)/cc-pVTZ and CCSDT(Q,fc)/cc-pVDZ calculations (ESI, Table 1S†). The energies of the reactions (1) and (2) are given as following −144.9 kJ mol−1 and −45.4 kJ mol−1 at the CCSDT(Q,fc)/cc-pVTZ theory level. The energy of the reaction (2a) was calculated to be 13.0 kJ mol−1 taking into account the energy of the reaction (1) obtained here and the experimental value of the O2(b1Σ+g) ← O2(X3Σ−g) transition energy. Thus, MeOOH is a high energetic molecule. Both pathways (2) and (2a) are energetically accessible because the decomposition of MeOOH with the O2(1Δg) elimination is weakly exothermic (Er = −45.4 kJ mol−1) while, in the generation of O2(b1Σ+g), the decomposition reaction is weakly endothermic (Er = 13.0 kJ mol−1).
Both theory levels (composite and multi-reference) are in agreement with the results of the relaxed scan of the Al–O(O) bond in 11 at the B3LYP/cc-pVTZ theory level: (1) there is no maximum on the potential curve, i.e., the decomposition of 11 on the singlet PES occurs with no transition state; (2) the barrier height is 19.0 kJ mol−1 (CCSD(T)//B3LYP with cc-pVTZ basis set), 34.7 kJ mol−1 (CASSCF). It is worthwhile to note that no intruder states were detected during the orbital update for the CASSCF calculation because the potential curve for the relaxed scan was smooth. The optimized structure of (MeO)3Al·O2 was not distorted, i.e., it was similar to the structure 11 optimized with the B3LYP method, but the Al–O-bond was longer. No η1-superoxo-η2-peroxo isomerism reported60–65 for the compounds of transition metals was revealed for (MeO)3Al·O2.
Next, to distinguish between the reactions (3) and (3a), the energy of the reaction (4) was calculated with the composite CCSD(T,fc)/cc-pVTZ//B3LYP/cc-pVTZ approach: Er is −104.6 (−115.1) kJ mol−1, the value for B3LYP is given in parentheses for comparison. One can conclude that the electronic configuration of the 3O2 molecule is correctly described by the CCSD(T) method (see the comparison of the results in ref. 58). Here, one can note that, for 11, the non-dynamic electron correlation is taken into account by the CCSD(T) method66 as was revealed by the T1 diagnostic (0.019) and the largest T2 amplitude with absolute value of 0.15 calculated at the CCSD(T)/6-31+G(2df,p) theory level. The use of the flexible cc-pVTZ basis set for coupled cluster method provides the result which is nearly free from the basis set incompleteness error. Thus, one can believe that the Er value of −104.6 kJ mol−1 is a reliable estimation.
However, for reactions with the (MeO)3Al·O2 intermediate, due to the especial time and resource consumption of calculations with the CCSDT(Q) method even with the moderate cc-pVDZ basis set, it is impossible to use this method for estimations with the focal point analysis. The energy of the intermediate 11 was calculated at the CCSD(T)/cc-pVTZ theory level as well as the energy of the reaction (4). On the basis of these results, the relative energy of the (MeO)3Al + O2(1Δg) level is estimated with the CCSDT(Q)/cc-pVTZ approach for the O2(1Δg)/O2(X3Σ−g) system. The relative energy of the (MeO)3Al + O2(b1Σ+g) level is obtained on the basis of the experimental energy of the excitation O2(b1Σ+g) ← O2(a1Δg) and the calculated energy of the reaction (3).
One can believe that the chosen combination of the CCSD(T) and CCSDT(Q) methods provides reliable results competing with the experimental data. Thus, the calculated reaction energies for (MeO)3Al·O2 and experimentally obtained values for the excitation O2(b1Σ+g) ← O2(1Δg) will be assembled on one energetic diagram (Fig. 8) and analyzed jointly. The level of isolated (MeO)3Al + O2(X3Σ−g) is selected as the initial point.
 | ||
| Fig. 8 Energetic levels of (MeO)3Al·O2 (the structure 11) and different asymptotics, i.e., (MeO)3Al + O2(3Σ−g), (MeO)3Al + O2(1Δg), (MeO)3Al + O2(1Σ+g), and quasi-isolated [(MeO)3Al + 1O2]. The (MeO)3Al·O2 and (MeO)3Al + O2(3Σ−g) levels were calculated with the CCSD(T)/cc-pVTZ//B3LYP/cc-pVTZ composite method; the energies of the levels (MeO)3Al + O2(3Σ−g) and (MeO)3Al + O2(1Δg) were estimated at the CCSDT(Q)/cc-pVTZ theory level, [(MeO)3Al + 1O2] was obtained by the relaxed scan at the CAS(14,10)/6-311G(d) theory level. The relative energies are in kJ mol−1. The dotted line indicates the relaxed scan. The arrows connect pair of energy levels calculated by the depicted computational procedure. The crossed arrows correspond to assuming reaction pathways which contradict with the obtained data. | ||
The main features of the determined location of the levels are: (1) the energy of the (MeO)3Al + O2(1Δg) system is lower than the level of (MeO)3Al·O2 (complex 11); (2) the quasi-isolated system [(MeO)3Al + 1O2] is energetically higher than 11; (3) the energy of the [(MeO)3Al + 1O2] coincides with the energy of (MeO)3Al + O2(b1Σ+g). Assuming that the (MeO)3Al·O2 dissociation corresponds to the (MeO)3Al + O2(1Δg), it is necessary to conclude that if the dissociation proceeds without a transition state (Fig. 8), the potential energy curve of the relaxed PES will exhibit decrease of the energy or if a saddle point or a conical intersection is located in the reaction pathway (Fig. 8), the curve will show an increase with subsequent decrease of the energy. However, as was calculated with quantum chemical methods, the energy curve smoothly increases, that is, the features of the relaxed scan curve for the assuming reactions are totally opposite. Therefore, one can conclude that the dissociation asymptotics of (MeO)3Al·O2 corresponds to (MeO)3Al + O2(b1Σ+g) rather than to (MeO)3Al + O2(1Δg).
The position of the η1-(MeO)3Al·OO level (Fig. 8) in the system of energy levels (MeO)3Al + O2(b1Σ+g), (MeO)3Al + O2(1Δg), and (MeO)3Al + O2(X3Σ−g) calculated with the CCSD(T) and CCSDT(Q) methods and the position of the quasi-isolated [(MeO)3Al + 1O2] level determined by the CASSCF method is of crucial importance for the generation of O2(b1Σ+g). The energy of chemical bonds of two MeOOH is transferred through η1-(MeO)3Al·OO into the energy of the excited O2 eliminated in the adiabatic decomposition reaction. The key factor to implement the quite unusual non-photoinduced process, i.e., the chemical excitation O2(b1Σ+g) ← O2(X3Σ−g), is the multi-pathway sequence of elementary reactions leading for the model system to (MeO)2AlOOOMe and, next, to η1-(MeO)3Al·OO. The (MeO)3Al·OO complex is the key intermediate for the O2(b1Σ+g) generation. On the basis of the conducted energy analysis for the diagram (Fig. 8), the hypothesis about the elimination of O2(b1Σ+g) in adiabatic reaction on the singlet PES and in the catalytic reaction of the MeOOH decomposition, where (MeO)3Al is the catalyst, can be considered to be justified with quantum chemical methods.
To our knowledge, the established in the present study the electronically excited state of the dioxygen, O2(b1Σ+g), eliminated in the catalytic reaction of the MeOOH decomposition (the model system) is the first report about the chemical excitation of O2 to the so-called second singlet state, O2(b1Σ+g). Here, it should be noted that the O2(1Δg) state is doubly degenerate. This phenomenon is new for the chemistry of peroxides and for catalysis on transition and non-transition metal centers. The catalytic oxidation of various substrates by the system t-BuOOH-transition metal compound67–73 conducted in the atmospheric O2, the activation of O2(X3Σ−g) and the formation of 1:1 adducts of catalyst with O2(1Δg) as well as the O2(1Δg) photoinduced elimination74,75 have been shown previously. While the reactivity of O2(1Δg) has been studied for about 80 years,76–80 the chemistry of O2(b1Σ+g) is not essentially defined. Indeed, firstly, the lifetime of O2(b1Σ+g) determined by the spin-allowed transition O2(b1Σ+g) → O2(1Δg)75,79,81,82 is considerably shorter than the lifetime of O2(1Δg), i.e., the quenching79,83–86 is quantitative. Second, O2(b1Σ+g) is quantitatively deactivated through the electron–vibrational (e–v) mechanism unambiguously determined in ref. 83, 84 and 87. Moreover, as was shown in the earlier studies,83,84,87 the oxidation of substrates by O2(b1Σ+g) with C–H bonds becomes evading in the presence of the highly effective pathway of quenching.
Probably, the η1-(MeO)3Al·OO complex can show reactivity which could mimic O2(b1Σ+g), but in reaction pathway with bypass of O2(b1Σ+g) quenching. The selective oxidation12,13 of C–H bonds by the reactive forms of oxygen generated on the atoms of non-transition metals is a promising alternative to the developed catalytic systems on transition metals.88
4.3. Non-adiabatic reactions in the MeOOH/(MeO)3Al system and the possibility of the chemically activated O2 formation
In order to find the possibility of “hot” O2(3Σ−g) generation in the strongly exothermic net reaction (1) and in the elimination reaction (4), which are not spin-conserving reactions, non-adiabatic reaction pathways, were searched and the triplet PES regions were studied with the B3LYP density functional. To determine electronic ground states for the located structures, the energies of the singlet–triplet vertical excitation of (MeO)3Al and the intermediates were calculated. Then, the local minima and the energy degeneracy points (Minimum Energy Crossing Point, MECP) of the singlet and triplet PESs were searched.The singlet spin coupling is the ground state for (MeO)3Al since, as was found at the B3LYP/cc-pVTZ theory level, the energy of the singlet–triplet transition (T1 ← S0) is (564.5 kJ mol−1). Thus, the asymptotics corresponding to the isolated monomers 3(MeO)3Al + 3O2 is not reachable from 11 with any remarkable thermodynamic probability. However, the singlet state is not the ground electronic state for 11 (Fig. 4), the triplet state is −79.0 kJ mol−1 more stable energetically than the lowest lying singlet state of the structure 11. Structure 16 (ESI, Fig. 1S†) is obtained in the geometry optimization of 11 (Fig. 4) on the triplet PES. The singlet state is the ground state for mono-η2-, bis-η2-methyl peroxides, and ozonides as was predicted at the B3LYP/cc-pVTZ theory level.
The search for the degeneracy of the singlet and triplet PESs for 3a, 7, 10, and 11 (Fig. 2 and 4) revealed minimum energy crossing points and, thus, non-stationary points of PES which determine shape of PES and non-adiabatic pathways for the net catalytic reaction. Structures corresponding to the MECPs are denoted as 3a-MECP, 7-MECP, 10-MECP, and 11-MECP (Fig. 9). Their energies are given: 98.5, 91.5, 2.4, and 10.5 kJ mol−1 at the B3LYP/cc-pVTZ theory level relative to 3a, 7, 10, and 11, respectively.
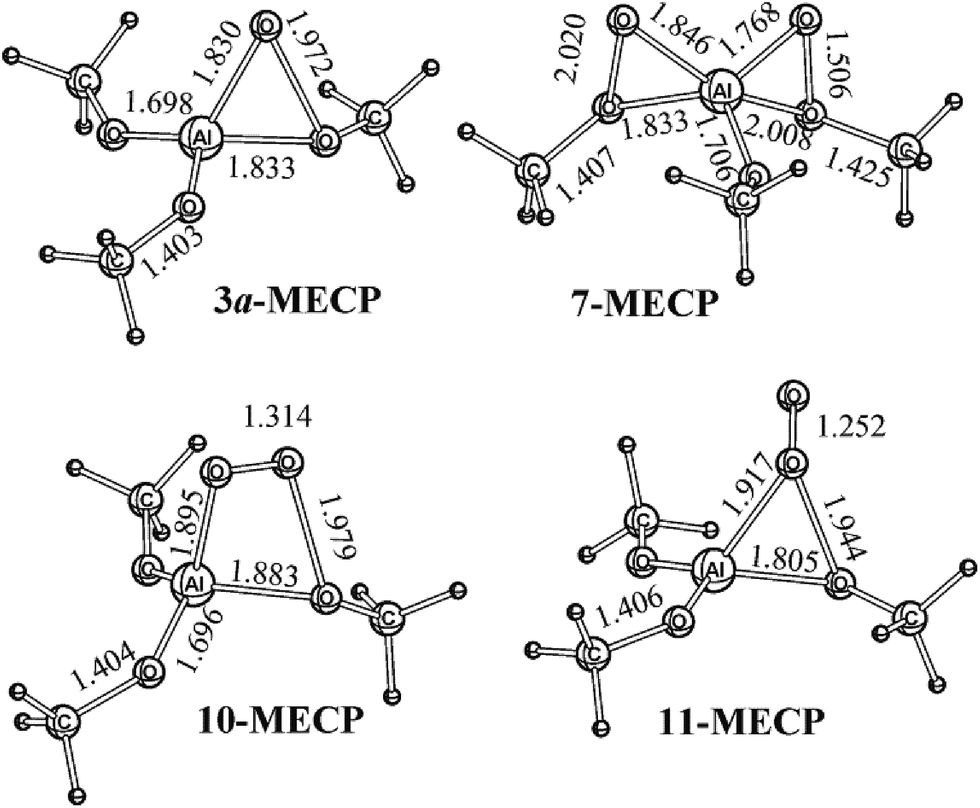 | ||
| Fig. 9 Structures corresponding to the minimum energy crossing points for the singlet and triplet PESs optimized at the B3LYP/cc-pVTZ theory level. Bond lengths are in Å. | ||
3a-MECP is a critical point which is not a stationary point of the singlet or triplet PESs for the AlOO cycle opening in 3a (Fig. 2) with the formation of structure 3b (Fig. 10). The structure 3b is 58.1 kJ mol−1 less stable than 3a (the singlet ground state). The decomposition of 3b with the formation of O(3P) and (MeO)3Al occurs with the reaction energy (Er) of 50.5 kJ mol−1. However, for the structure with two AlOO cycles, 7, the singlet–triplet spin recoupling does not lead to the cycle opening, i.e., in 7b (Fig. 10) with the energy of 75.8 kJ mol−1 above the level of 7 (Fig. 1) was located. The O–O bond lengths optimized at the B3LYP/cc-pVTZ theory level are 2.301 Å and 1.501 Å while in 7 the bonds are of 2.020 Å and 1.506 Å. Spin conversions via 10-MECP (Ea,T←S = 2.4 kJ mol−1) and 11-MECP (Ea,T←S = 10.5 kJ mol−1) result in the strongly exothermic decomposition reaction 11 → (MeO)3Al + 3O2 (Er = −119.8 kJ mol−1). The stabilization of weakly bound complex 16 (ESI, Fig. 1S†) is unlikely under such conditions and the reaction leads to the elimination of the (MeO)3Al and  monomers. The asterisk denotes here and below a dioxygen molecule with the excess in energy. In this case, the O2 molecule is in the ground electronic state, but it is also vibrationally and/or translationally excited, i.e.,
monomers. The asterisk denotes here and below a dioxygen molecule with the excess in energy. In this case, the O2 molecule is in the ground electronic state, but it is also vibrationally and/or translationally excited, i.e.,  can show higher or probably uncommon reactivity as compared to O2 thermalized under the (mild) conditions of the synthesis.12,13,89 Moreover, these non-adiabatic reactions show the possibility of incomplete conversion of 11 in (MeO)3Al and O2(b1Σ+g); thus, in addition to physical quenching in O2(b1Σ+g) → O2(1Δg) and O2(1Δg) → O2(X3Σ−g), it is a reaction pathway without the generation of 1O2. It agrees with the experimentally determined13 incomplete (about 50%) conversion of dioxygen in the reaction with singlet dioxygen trap, 9,10-methylanthracene, in weakly quenching solvent. The probability of low-energy singlet–triplet transition (the activation energies Ea,T←S(10-MECP) = 2.4 kJ mol−1 and Ea,T←S(11-MECP) = 10.5 kJ mol−1, and Ea(11 → (MeO)3Al + O2(b1Σ+g)) = 53.3 kJ mol−1 are given for comparison) makes it possible to assume the possibility of the process controlling by selection of the solvent, i.e., stimulation of the adiabatic decomposition reaction and, thus, increase of the O2(b1Σ+g) yield.
can show higher or probably uncommon reactivity as compared to O2 thermalized under the (mild) conditions of the synthesis.12,13,89 Moreover, these non-adiabatic reactions show the possibility of incomplete conversion of 11 in (MeO)3Al and O2(b1Σ+g); thus, in addition to physical quenching in O2(b1Σ+g) → O2(1Δg) and O2(1Δg) → O2(X3Σ−g), it is a reaction pathway without the generation of 1O2. It agrees with the experimentally determined13 incomplete (about 50%) conversion of dioxygen in the reaction with singlet dioxygen trap, 9,10-methylanthracene, in weakly quenching solvent. The probability of low-energy singlet–triplet transition (the activation energies Ea,T←S(10-MECP) = 2.4 kJ mol−1 and Ea,T←S(11-MECP) = 10.5 kJ mol−1, and Ea(11 → (MeO)3Al + O2(b1Σ+g)) = 53.3 kJ mol−1 are given for comparison) makes it possible to assume the possibility of the process controlling by selection of the solvent, i.e., stimulation of the adiabatic decomposition reaction and, thus, increase of the O2(b1Σ+g) yield.
 | ||
| Fig. 10 Structures for triplet PES optimized at the B3LYP/cc-pVTZ theory level. Bond lengths are in Å. | ||
The reaction η1-(MeO)3Al·OO → (MeO)3Al + O2 is a neither single-pathway nor product spin-pure process, so it is possible that the reactivity of O2(b1Σ+g)/O2(1Δg) (the latter one is the product of the spin-allowed quenching of O2(b1Σ+g)) and chemically activated (translationally–vibrationally–rotationally hot) 3O2, which is the product of the spin-forbidden reaction, may be observed for the t-BuOOH/(t-BuO)3Al system of experimental interest.
5. Conclusion
On the basis of the quantum chemical study of the MeOOH/(MeO)3Al model system, it was exhibited for the first time that O2 is generated chemically excited. One can conclude that the excited dioxygen, O2(b1Σ+g), is produced in the liquid phase in the catalytic decomposition of t-BuOOH. O2(b1Σ+g) is formed as a result of the successive reactions of aluminum tert-butoxide with tert-butyl hydroperoxide at the mole ratio of 1:2. In the first step, the aluminum-containing peroxide (t-BuO)2AlOOt-Bu for the experimental system or (MeO)2AlOOMe for the model system is produced. The latter one reacts with the second MeOOH molecule both through the pathway of the substitution of the methoxy group (main reaction pathway) and through the six-membered transition state (minor pathway) with the formation of metastable metal-containing mono-η2- and bis-η2-methyl peroxides and the elimination of the MeOH molecule. The formation of the isomeric ozonides (MeO)2AlO3Me is the rate-determining step. For the main reaction pathway, the activation energy (Ea) of the intermolecular O atom transfer was calculated to be 159.2 kJ mol−1. From ozonide-1,3, (MeO)2AlOOOMe, the corresponding metal dioxygen intermediate, η1-(MeO)3Al·O2, is generated in the decomposition reaction with the elimination of dioxygen in the electron-excited state, O2(1Σg). The electronic state of O2 was established by the analysis of the energy diagram calculated for the (MeO)3Al/O2 system at the CCSD(T,fc)/cc-pVTZ, CCSDT(Q)/cc-pVTZ, and CAS(14,10)/6-311G(d) theory levels. For the decomposition of η1-(MeO)3Al·O2, a non-adiabatic pathway, that includes the singlet–triplet transition and leads to the generation of chemically activated 3O2, was exhibited.
The conducted simulation makes it possible to propose that the catalytic decomposition of t-BuOOH leads to a complicated oxidizer system being the source of the reactive oxygen species: mono-η2-, bis-η2-peroxides, isomeric ozonides, η1-peroxide, chemically activated  and chemically excited (O2(b1Σ+g) and O2(1Δg)) dioxygen. O2(1Δg) is the product of the decay of the higher excited state. O2(b1Σ+g) is characterized by the significantly shorter lifetime than O2(1Δg). The latter is the product of the spin-allowed transition decomposition of O2(b1Σ+g). One can believe that quenching through the e–v mechanism for O2(b1Σ+g) can create a pathway to exhibit an uncommon reactivity of O2(1Δg) which results from the chemical activation of the substrate by energy excess released in the O2(1Δg) ← O2(b1Σ+g) relaxation. The formation of the (t-BuO)3Al·O2 complex analogous to (MeO)3Al·O2 predicted here on the basis of quantum chemical study can determine the reactivity of chemically immobilized O2(b1Σ+g). Both pathways make the new chemistry of 1O2 possible, that is, the oxidizing activity results directly or indirectly from the reactivity of O2(b1Σ+g). Thus, it may determine important enhancement to famous Fenton and Haber–Weiss chemistry.
and chemically excited (O2(b1Σ+g) and O2(1Δg)) dioxygen. O2(1Δg) is the product of the decay of the higher excited state. O2(b1Σ+g) is characterized by the significantly shorter lifetime than O2(1Δg). The latter is the product of the spin-allowed transition decomposition of O2(b1Σ+g). One can believe that quenching through the e–v mechanism for O2(b1Σ+g) can create a pathway to exhibit an uncommon reactivity of O2(1Δg) which results from the chemical activation of the substrate by energy excess released in the O2(1Δg) ← O2(b1Σ+g) relaxation. The formation of the (t-BuO)3Al·O2 complex analogous to (MeO)3Al·O2 predicted here on the basis of quantum chemical study can determine the reactivity of chemically immobilized O2(b1Σ+g). Both pathways make the new chemistry of 1O2 possible, that is, the oxidizing activity results directly or indirectly from the reactivity of O2(b1Σ+g). Thus, it may determine important enhancement to famous Fenton and Haber–Weiss chemistry.
Acknowledgements
A. I. P. is thankful to SFU Super - computer Centre for generous donation of computer time. O. B. G. is thankful for the partial support of this study provided by a grant under agreement between N.I. Lobachevsky State University of Nizhny Novgorod and the Ministry of Education and Science of the Russian Federation.References
- K. Weissermel and H.-J. Arpe, in Industrial Organic Chemistry, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2008, pp. 193–215 Search PubMed.
- J. Lewiński, J. Zachara, P. Goś, E. Grabska, T. Kopeć, I. Madura, W. Marciniak and I. Prowotorow, Chem.–Eur. J., 2000, 6, 3215–3227 CrossRef.
- J. Lewiński and A. H. Wheatley, in Modern Organoaluminum Reagents, ed. S. Woodward and S. Dagorne, Springer, Berlin-Heidelberg, Germany, 2013, vol. 41, ch. 55, pp. 1–58 Search PubMed.
- G. A. Tolstikov, S. K. Minsker, R. G. Bulgakov, G. Y. Maistrenko, A. V. Kuchin, U. M. Dzhemilev and V. P. Kazakov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1979, 28, 2471 CrossRef.
- V. A. Dodonov, L. A. Stepovik and S. M. Safonova, Russ. J. Gen. Chem., 1990, 60, 1839–1848 CAS.
- V. A. Dodonov, L. A. Stepovik, S. M. Safonova and V. A. Zinchenkov, Russ. J. Gen. Chem., 1990, 60, 1125–1136 CAS.
- V. A. Dodonov, Bulletin of Lobachevsky Nyzhniy Novgorod State University, 2012, 2, 81–101 Search PubMed.
- L. P. Stepovik, I. M. Martinova, V. A. Dodonov and V. K. Cherkasov, Russ. Chem. Bull., 2002, 51, 638–644 CrossRef CAS.
- V. A. Dodonov, L. P. Stepovik, A. C. Soskova and E. A. Zaburdaeva, Russ. J. Gen. Chem., 1994, 6, 1715–1721 Search PubMed.
- L. P. Stepovik, V. A. Dodonov and E. A. Zaburdaeva, Russ. J. Gen. Chem., 1997, 67, 116–120 Search PubMed.
- V. A. Dodonov, E. A. Zaburdaeva, N. B. Dolganova, L. P. Stepovik and T. I. Zinov'eva, Russ. J. Gen. Chem., 1997, 67, 988–992 Search PubMed.
- V. A. Dodonov, E. A. Zaburdaeva and L. P. Stepovik, Russ. Chem. Bull., 2004, 53, 1729–1734 CrossRef CAS.
- E. A. Zaburdaeva, V. A. Dodonov and L. P. Stepovik, J. Organomet. Chem., 2007, 692, 1265–1268 CrossRef CAS.
- E. A. Zaburdaeva and V. A. Dodonov, Russ. Chem. Bull., 2011, 1, 177–179 Search PubMed.
- F. Naso, M. A. M. Capozzi, A. Bottoni, M. Calvaresi, V. Bertolasi, F. Capitelli and C. Cardellicchio, Chem.–Eur. J., 2009, 15, 13417–13426 CrossRef CAS PubMed.
- M. A. M. Capozzi, C. Centrone, G. Fracchiolla, F. Naso and C. Cardellicchio, Eur. J. Org. Chem., 2011, 4327–4334 CrossRef CAS.
- G. E. O'Mahony, A. Ford and A. R. Maguire, J. Sulfur Chem., 2012, 34, 301–341 CrossRef.
- C. Bolm, O. Beckmann, T. Kühn, C. Palazzi, W. Adam, P. B. Rao and C. R. Saha-Möller, Tetrahedron: Asymmetry, 2001, 12, 2441–2446 CrossRef CAS.
- C. Bolm, O. Beckmann and C. Palazzi, Can. J. Chem., 2001, 79, 1593–1597 CrossRef CAS.
- T. Yamaguchi, K. Matsumoto, B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2007, 46, 4729–4731 CrossRef CAS PubMed.
- K. Matsumoto, T. Yamaguchi and T. Katsuki, Chem. Commun., 2008, 1704–1706 RSC.
- C. S. Foote, Acc. Chem. Res., 1968, 1, 104–110 CrossRef CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- W. Adam, D. V. Kazakov and V. P. Kazakov, Chem. Rev., 2005, 105, 3371–3387 CrossRef CAS PubMed.
- S. Miyamoto, G. E. Ronsein, F. M. Prado, M. Uemi, T. C. Corrêa, I. N. Toma, A. Bertolucci, M. C. B. Oliveira, F. D. Motta, M. H. G. Medeiros and P. D. Mascio, IUBMB Life, 2007, 59, 322–331 CrossRef CAS PubMed.
- A. A. Frimer, Singlet oxygen, CRC Press, Boca Raton, 1985 Search PubMed.
- H. H. Wasserman and R. W. Murray, Singlet oxygen, Academic Press, New York, 1979 Search PubMed.
- B. Halliwell, Am. J. Med., 1991, 91, S14–S22 CrossRef.
- B. Halliwell and J. Gutteridge, in Free Radicals in Biology and Medicine, Clarendon Press, Oxford, England, United Kingdom, 1985, pp. 279–315 Search PubMed.
- A. Bast, G. R. M. M. Haenen and C. J. A. Doelman, Am. J. Med., 1991, 91, S2–S13 CrossRef.
- I. E. Kochevar and R. W. Redmond, in Methods in Enzymology, ed. L. Packer and H. Sies, Academic Press, 2000, vol. 319, pp. 20–28 Search PubMed.
- C. Pierlot, J.-M. Aubry, K. Briviba, H. Sies and P. D. Mascio, in Methods in Enzymology, ed. L. Packer and H. Sies, Academic Press, New York, 2000, vol. 319, pp. 3–20 Search PubMed.
- F. Munoz, E. Mvula, S. E. Braslavsky and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 2001, 1109–1116 RSC.
- G. Merényi, J. Lind, S. Naumov and C. von Sonntag, Chem.–Eur. J., 2010, 16, 1372–1377 CrossRef PubMed.
- S. Naumov, G. Mark, A. Jarocki and C. von Sonntag, Ozone: Sci. Eng., 2010, 32, 430–434 CrossRef CAS.
- R. H. Hertwig and W. Koch, Chem. Phys. Lett., 1997, 268, 345–351 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 03, Version D.02, Gaussian, Inc, Wallingford, CT, 2007 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- B. Bolding and K. Baldridge, Comput. Phys. Commun., 2000, 128, 55–66 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, in Theory and Applications of Computational Chemistry, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, The Netherlands, 2005, pp. 1167–1189 Search PubMed.
- J. L. Bentz, R. M. Olson, M. S. Gordon, M. W. Schmidt and R. A. Kendall, Comput. Phys. Commun., 2007, 176, 589–600 CrossRef CAS.
- P. Piecuch, S. A. Kucharski, K. Kowalski and M. Musiał, Comput. Phys. Commun., 2002, 149, 71–96 CrossRef CAS.
- J. F. Stanton, J. Gauss, M. E. Harding and P. G. Szalay, CFOUR (Coupled-Cluster techniques for Computational Chemistry) with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, Y. J. Bomble, L. Cheng, O. Christiansen, M. Heckert, O. Heun, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, K. Klein, W. J. Lauderdale, D. A. Matthews, T. Metzroth, D. P. O'Neill, D. R. Price, E. Prochnow, K. Ruud, F. Schiffmann, W. Schwalbach, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J. D. Watts and the integral packages MOLECULE (J. Almlöf and P. R. Taylor) PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. Aa. Jensen, P. Jørgensen and J. Olsen) and ECP routines by A. V. Mitin and C. van Wüllen, URL: http://slater.chemie.uni-mainz.de/cfour/index.php?n=Main.HomePage, accessed August 26th, 2016.
- M. Kállay and P. R. Surján, J. Chem. Phys., 2001, 115, 2945–2954 CrossRef.
- M. Kállay and J. Gauss, J. Chem. Phys., 2008, 129, 144101 CrossRef PubMed.
- M. Kállay, Z. Rolik, J. Csontos, I. Ladjánszki, L. Szegedy, B. Ladóczki, G. Samu, K. Petrov, M. Farkas, P. Nagy, D. Mester and B. Hégely, Mrcc, a quantum chemical program suite, URL: http://www.mrcc.hu, accessed August 26th, 2016.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95–99 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, ORCA-An Ab Initio, DFT and Semiempirical electronic structure package, version 3.0.3, Max-Planck-Institute for Chemical EnergyConversion Stiftstr, Mülheim a. d. Ruhr, Germany, 34-36, 45470, accessed August 10th, 2016 Search PubMed.
- S. K. Ignatov, MOLTRAN: a program for molecular visualization and thermodynamic calculations, N. I. Lobachevsky State University of Nizhny Novgorod, Nizhny Novgorod, 2009, URL: http://www.unn.ru/chem/moltran/, accessed August 26th, 2016 Search PubMed.
- R. Dennington Roy II, K. Todd, M. John, E. Ken, H. W. Lee and G. Ray, GaussView, Version 03, Semichem Inc, Shawnee Mission, KS, 2003 Search PubMed.
- G. Zhurko, ChemCraft, Version 1.7 (build 365), URL: http://www.chemcraftprog.com, accessed July 20th, 2016 Search PubMed.
- O. B. Gadzhiev, S. K. Ignatov, M. Y. Kulikov, A. M. Feigin, A. G. Razuvaev, P. G. Sennikov and O. Schrems, J. Chem. Theory Comput., 2012, 9, 247–262 CrossRef PubMed.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure IV. Constants of Diatomic Molecules, Van Nostrand Reinhold Co, New York, 1979 Search PubMed.
- Y. Gong, M. Zhou and L. Andrews, Chem. Rev., 2009, 109, 6765–6808 CrossRef CAS PubMed.
- C. J. Cramer and W. B. Tolman, Acc. Chem. Res., 2007, 40, 601–608 CrossRef CAS PubMed.
- D. M. Wagnerová and K. Lang, Coord. Chem. Rev., 2011, 255, 2904–2911 CrossRef.
- A. Yokoyama, J. E. Han, J. Cho, M. Kubo, T. Ogura, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2012, 134, 15269–15272 CrossRef CAS PubMed.
- J. Cho, H. Y. Kang, L. V. Liu, R. Sarangi, E. I. Solomon and W. Nam, Chem. Sci., 2013, 4, 1502–1508 RSC.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- D. G. Liakos and F. Neese, J. Chem. Theory Comput., 2011, 7, 1511–1523 CrossRef CAS PubMed.
- T. Matsushita, D. T. Sawyer and A. Sobkowiak, J. Mol. Catal. A: Chem., 1999, 137, 127–133 CrossRef CAS.
- A. Sobkowiak, A. Qui, X. Liu, A. Llobet and D. T. Sawyer, J. Am. Chem. Soc., 1993, 115, 609–614 CrossRef CAS.
- D. T. Sawyer, A. Sobkowiak and T. Matsushita, Acc. Chem. Res., 1996, 29, 409–416 CrossRef CAS.
- D. A. Pantazis and J. E. McGrady, Inorg. Chem., 2003, 42, 7734–7736 CrossRef CAS PubMed.
- D. T. Sawyer, C. Sheu, H.-C. Tung and A. Sobkowiak, in Studies in Surface Science and Catalysis, ed. L. I. Simándi, Elsevier, Amsterdam, The Netherlands, 1991, vol. 66, pp. 285–296 Search PubMed.
- C. Kang, C. Redman, V. Cepak and D. T. Sawyer, Bioorg. Med. Chem., 1993, 1, 125–140 CrossRef CAS PubMed.
- M. Drees, S. A. Hauser, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2013, 748, 36–45 CrossRef CAS.
- A. A. Abdel-Shafi, M. D. Ward and R. Schmidt, Dalton Trans., 2007, 2517–2527 RSC.
- R. Schmidt, Photochem. Photobiol., 2006, 82, 1161–1177 CrossRef CAS PubMed.
- P. R. Ogilby, Photochem. Photobiol. Sci., 2010, 9, 1543–1560 CAS.
- A. A. Frimer, in Peroxides, ed. S. Patai, John Wiley & Sons, Ltd, Chichester, England, United Kingdom, 1983, pp. 201–234 Search PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1758 CrossRef CAS PubMed.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- R. Schmidt, J. Phys. Chem. A, 2006, 110, 5990–5997 CrossRef CAS PubMed.
- D. Weldor, T. D. Poulsen, K. V. Mikkelsen and P. R. Ogilby, Photochem. Photobiol., 1999, 70, 369–379 CrossRef.
- R. Schmidt, J. Photochem. Photobiol., A, 1994, 80, 1–5 CrossRef CAS.
- M. Bodesheim and R. Schmidt, J. Phys. Chem. A, 1997, 101, 5672–5677 CrossRef CAS.
- R. Schmidt and M. Bodesheim, J. Phys. Chem. A, 1998, 102, 4769–4774 CrossRef CAS.
- R. D. Scurlock, B. Wang and P. R. Ogilby, J. Am. Chem. Soc., 1996, 118, 388–392 CrossRef CAS.
- R. Schmidt and M. Bodesheim, J. Phys. Chem., 1994, 98, 2874–2876 CrossRef CAS.
- E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W.-D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S.-F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- V. A. Dodonov, E. A. Zaburdaeva and L. P. Stepovik, Russ. J. Gen. Chem., 2000, 70, 1482–1483 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized Cartesian coordinates for reported intermediates, transition states, and minimum energy crossing points; the data for the focal point analysis; the reaction pathways for the peroxides dissociation, and additional comments on chemical reactivity of the catalytic system. See DOI: 10.1039/c6ra21471a |
| This journal is © The Royal Society of Chemistry 2016 |