DOI:
10.1039/C6RA21403G
(Paper)
RSC Adv., 2016,
6, 110255-110265
Two fluorescent lead phosphonates for highly selective sensing of nitroaromatics (NACs), Fe3+ and MnO4− ions†
Received
26th August 2016
, Accepted 3rd November 2016
First published on 4th November 2016
Abstract
Two novel lead(II) phosphonates with a 2D double-layer and a 3D framework structure, namely, [Pb(BPDP)] (1) and [Pb3(BPDP)1.5(OOCC6H4COOH)3] (2) (H2BPDP = 4,4′-biphenyldiphosphonate(monoethyl ester), H2bdc = HOOCC6H4COOH), have been synthesized under hydrothermal conditions. Compounds 1 and 2 are stable in air and insoluble in water. Thermogravimetric analyses reveal that compounds 1 and 2 remain unchanged up to about 233 °C and 212 °C, respectively. Luminescence explorations demonstrated that compounds 1 and 2 exhibit highly selective and sensitive sensing for nitroaromatics (NACs). In ion recognition, the choice of solvent for compounds 1 and 2 has a different effect. Experiments proved that the identification of compounds 1 and 2 in aqueous solution is superior to that in ethanol solution. Moreover, compounds 1 and 2 also exhibit highly selective and sensitive sensing for Fe3+ and MnO4−. These results reveal that compounds 1 and 2 may be excellent fluorescent sensors for p-NP, Fe3+, and MnO4−.
Introduction
MOF materials, due to their luminosity, have a potential application in the detection of a variety of analytes, such as nitroaromatics (NACs), cations, anions, biomolecules, small molecules, and volatile organic compounds.1,2 Selective and sensitive detection of nitroaromatics (NACs) is of current interest for a variety of reasons, including national security and environmental concerns.3 The detection of these life-threatening energetic materials plays a crucial role in anti-terrorism operations, homeland security, and civilian safety, and thus has the ability to directly save human lives and protect the environment.4 Therefore, a highly sensitive detection of NACs has become a critical issue. However, most of these methods offer suffer from a number of drawbacks, such as high cost and poor portability, which prevent their widespread application.
Fluorescence-quenching-based sensing has emerged as a new approach that is cost effective, fast and easily portable.5 For this purpose, some luminescent MOFs have been tested for their potential applications in the detection of electron-deficient nitroaromatics by the fluorescence quenching method.6 In our daily life, iron is becoming more and more indispensable and important. At the same time, iron is a ubiquitous element in the human body, which also plays an important role in all the biological systems. The Fe3+ ion influences electron transfer and haemoglobin formation.7 Both an excess and a deficiency are harmful and may cause serious diseases, such as anemia, heart failure, damage to nucleic acids and proteins, cancer, etc.8 As a consequence, the selective detection of Fe3+ is very important for human health. At present, only a few fluorescent sensors that sense Fe3+ and Cu2+ ions have been reported.9 To date, the selective detection of Fe3+ has not been reported by our group. In recent decades, water pollution has become a global environmental issue together with the detection of pollutants that have been found in industrial wastewater. In industrial applications, strong oxidizers have been widely used in bleaching agents, disinfectants, gas absorption agents, and CO2 extractants. In the research laboratory, acidic MnO4− solution is an important oxidation–reduction reagent.10 With the rapid development of industrialization, anionic pollutants have caused serious environmental problems. Moreover, an excessive MnO4− is harmful to human health, and it can cause gastrorrhagia, liver and kidney damage, and even deformity.11 Therefore, the selective detection of MnO4− is very important to our life environment. Recently, there has been increasing interest in metal phosphonates, which are a special family of metal–organic frameworks (MOFs), because of their enormous variety of interesting structural topologies and wide potential applications.12 To date, research on the properties of metal(II) phosphonates is mainly focused on their magnetism, luminescence, proton conductivity, and ion exchange, while there are few reports about ions and the molecular recognition properties of these materials. In recent years, the sensing of ions and small molecules utilizing some luminescent MOF materials has been realized.13 However, only a few metal phosphonate hybrids with molecular recognition properties have been obtained by our group.14 In this study, by employing 4,4′-biphenyldiphosphonate(monoethyl ester) (H2BPDP) as the phosphonate ligand and 1,4-benzenedicarboxylic acid (H2bdc) as the second metal linker, we have successfully obtained two novel lead phosphonates with a 2D double-layer and a 3D framework structure, namely, [Pb(BPDP)] (1), and [Pb3(BPDP)1.5(OOCC6H4COOH)3] (2). As far as we all know, this is the first time that H2bdc has been introduced to construct a metal(II) phosphonate assembled from 4,4′-biphenyldiphosphonate(monoethyl ester). Herein, we describe their synthesis, crystal structures, and luminescent and selective fluorescent sensing properties of NACs, and Fe3+ and MnO4− ions.
Experimental section
Materials and methods
The 4,4′-biphenyldiphosphonate(monoethyl ester) (H2BPDP) was obtained as described in the literature.15 All other chemicals were used as obtained without further purification. C and H were determined by using a PE-2400 elemental analyzer. P and Pb were determined by using an inductively coupled plasma (ICP) atomic absorption spectrometer. IR spectra were recorded on a Bruker AXS TENSOR-27 FT-IR spectrometer with KBr pellets in the range 4000–400 cm−1. The X-ray powder diffraction data was collected on a Bruker AXS D8 Advance diffractometer using Cu-Kα radiation (λ = 1.5418 Å) in the 2θ range 5–60° with a step size of 0.02° and a scanning rate of 3° min−1. TG analyses were performed on a Perkin-Elmer Pyris Diamond TG-DTA thermal analysis system in static air with a heating rate of 10 K min−1 from 50 °C to 1100 °C. The luminescence spectra were investigated on a HITACHI F-7000 spectrofluorometer. The metal content was measured by ICP atomic emission spectrometric analysis. UV-vis spectroscopic studies were performed on a Lambda 35 spectrophotometer.
Synthesis of [Pb(BPDP)] (1). A mixture of Pb(Ac)2·3H2O (0.19 g, 0.5 mmol) and H2BPDP (0.1 g, 0.25 mmol) was dissolved in 10 mL deionized water. The resulting solution was stirred for about 1 h at room temperature, sealed in a 20 mL Teflon-lined stainless steel autoclave, and heated at 140 °C for 3 d under autogenous pressure. Crystals of 1 were isolated in ca. 42.5% yield based on lead after cooling the Teflon reactor to room temperature. Anal. calcd for C18H22O6P2Pb: C, 35.82; H, 3.67; P, 10.26; Pb, 34.33%. Exp: C, 35.78; H, 3.63; P, 10.31; Pb, 34.38%. IR (KBr, cm−1): 3440 (w), 3070 (m), 2840 (w), 1648 (m), 1590 (m), 1535 (m), 1407 (s), 1095 (s), 1001 (s), 935 (m), 765 (m), 574 (m), 514 (m), 440 (m).
Synthesis of [Pb3(BPDP)1.5(OOCC6H4COOH)3] (2). A mixture of Pb(Ac)2·3H2O (0.19 g, 0.5 mmol), H2BPDP (0.1 g, 0.25 mmol), and H2bdc (0.04 g, 0.25 mmol) was dissolved in 10.0 mL of deionized water. The resulting solution was stirred for about 1 h at room temperature, sealed in a 20 mL Teflon-lined stainless steel autoclave, and heated at 140 °C for 3 d under autogenous pressure. After the mixture was cooled slowly to room temperature, colorless stick crystals of 2 were obtained in ca. 48.8% yield based on lead. Anal. calcd for C51H48O21P3Pb3: C, 35.79; H, 2.83; P, 5.43; Pb, 36.32%. Exp: C, 35.75; H, 2.86; P, 5.48; Pb, 36.28%. IR (KBr, cm−1): 3432 (m), 2977 (w), 2921 (w), 1684 (m), 1582 (m), 1541 (m), 1395 (s), 1283 (w), 1129 (s), 1037 (s), 956 (m), 833 (w), 782 (w), 741 (m), 598 (w), 526 (m).
X-ray crystallography
Data collections for compounds 1 and 2 were performed on a Bruker AXS Smart APEX II CCD X-diffractometer equipped with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 ± 2 K. An empirical absorption correction was applied using the SADABS program. The structures were solved by direct methods and refined by full matrix least-squares on F2 by using the program SHELXL-2014/7.16 All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms of organic ligands were generated geometrically with fixed isotropic thermal parameters, and included in the structure factor calculations. Details of crystallographic data and structural refinements of compounds 1 and 2 are summarized in Table 1. Selected bond distances and angles of compounds 1 and 2 are given in Tables S1 and S2.†
Table 1 Crystal data and structural refinements for compounds 1 and 2
| R1 = Σ(|F0| − |FC|)/Σ|F0|. wR2 = [Σw(|F0| − |FC|)2/ΣwF02]1/2. |
| Compounds |
1 |
2 |
| Empirical formula |
C18H22O6P2Pb |
C51H47O21P3Pb3 |
| Fw |
603.48 |
1710.36 |
| Temperature (K) |
295(2) |
295(2) |
| Crystal system |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a (Å) |
5.3295(4) |
12.1420(5) |
| b (Å) |
14.0859(9) |
15.4695(6) |
| c (Å) |
15.0058(10) |
15.6819(6) |
| α (deg) |
66.0950(10) |
96.6760(10) |
| β (deg) |
86.3290(10) |
94.1210(10) |
| γ (deg) |
81.2840(10) |
111.1240(10) |
| V (Å3) |
1017.97(12) |
2708.33(19) |
| Z |
2 |
2 |
| ρcalcd (g cm−3) |
1.969 |
2.097 |
| μ (mm−1) |
8.475 |
9.469 |
| Unique date, Rint |
5776, 0.0181 |
15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564, 0.0230 564, 0.0230 |
| Crystal size (mm) |
0.063 × 0.026 × 0.021 |
0.070 × 0.030 × 0.030 |
| Completeness to theta = 26.49 |
99.0% |
99.2% |
| GOF on F2 |
1.031 |
1.023 |
| R1a, wR2b (I > 2σ(l)) |
0.0285, 0.0693 |
0.0366, 0.0796 |
| R1a, wR2b (all data) |
0.0344, 0.0722 |
0.0573, 0.0872 |
Results and discussion
Syntheses
Hydrothermal reactions of H2BPDP with the corresponding Pb(Ac)2·3H2O in a 2:1 molar ratio at their original pH (T = 140 °C, 3 d) resulted in large single stick crystals of compound 1. Larger crystals and the best crystallinity for compound 2 were acquired with a molar ratio of Pb2+/H2BPDP/H2bdc = 2:1:1 at their original pH (T = 140 °C, 3 d). In order to obtain relatively pure phases of compounds 1 and 2, the reactions were carried out by changing the lead salts (Pb(NO3)2, PbCl2, and Pb(Ac)2·3H2O). It was found that lead salts play a crucial role in the formation of the reaction products. Pb(NO3)2 and PbCl2, acting as reactants, produce amorphous powders. However, mixed phases for compounds 1 and 2 are obtained by using Pb(Ac)2·3H2O. It was found that the reaction temperature also plays a significant role in the cultivation of high quality single crystals. Pure phases of compounds 1 and 2 were obtained at a reaction temperature of 140 °C, as confirmed by the powder XRD patterns (Fig. S1 and S2, ESI†). At T > 140 °C or T < 140 °C, no single-crystals formed.
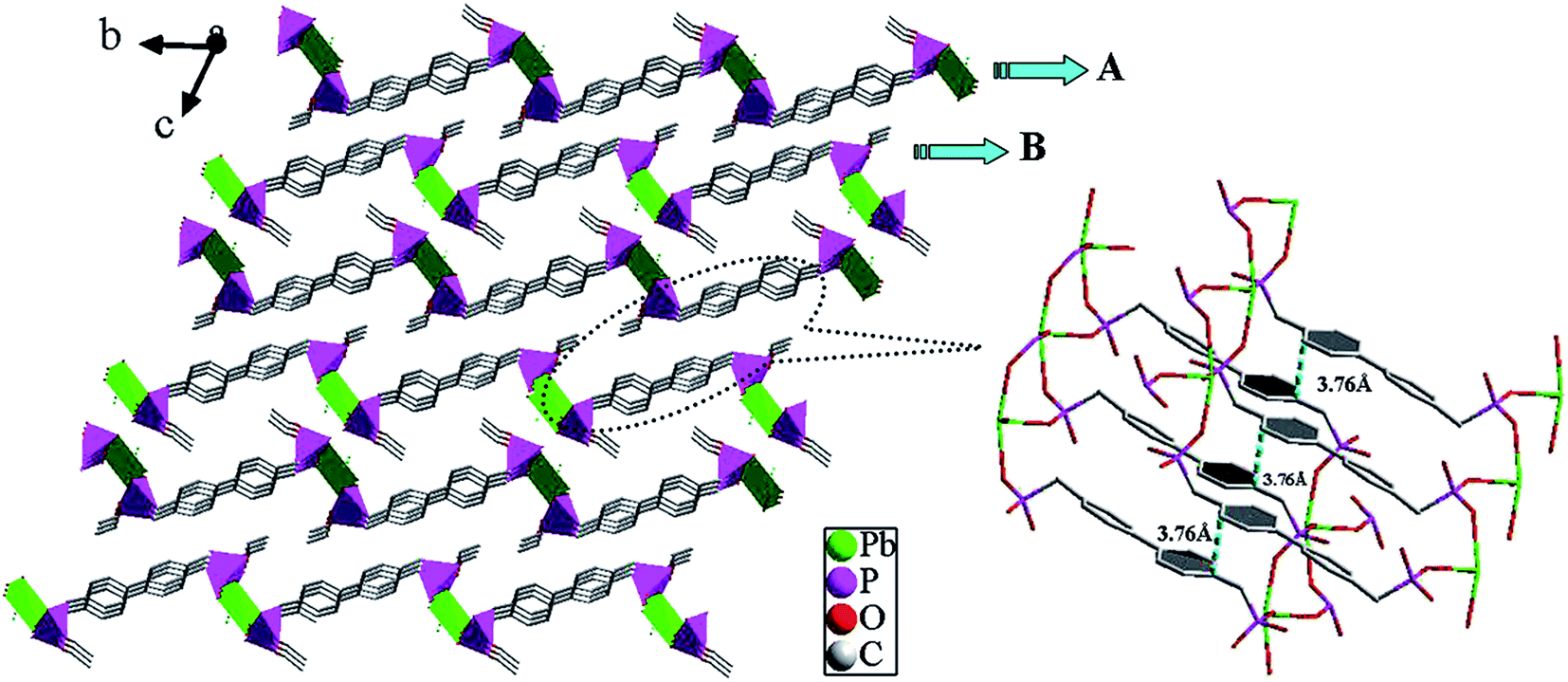
Crystal structures of [Pb(BPDP)] (1). Single crystal X-ray diffraction analysis proves that compound 1 crystallizes in the Triclinic P space group (see Table 1). As shown in Fig. 1, the asymmetric unit of 1 consists of a Pb(II) ion and a BPDP2− ion. The Pb(II) ion is four-coordinated by four oxygen atoms (O1A, O2B, O4, and O5C) from four phosphonate ligands, leading to a distorted tetrahedral geometry. The Pb–O bond lengths are in the range 2.290(4) to 2.578(4) Å, which is comparable to those reported for other lead(II) phosphonates.17 The coordination environment of the phosphonate ligand is displayed in Fig. 2a. In the structure, the BPDP2− anion is a pentadentate ligand, binding five separate Pb(1) ions through four O atoms (O2, O3, O4, and O5). The {PbO4} distorted tetrahedron and {CPO3} tetrahedra form a 2D inorganic layer in the ab-plane via corner-sharing (Fig. S3a, ESI†). The result of these connections is the formation of regular windows made up of 30 atoms, which consist of two Pb, four P, four O, and twenty C atoms with the sequence (Pb1–O2–P1–C1–C2–C3–C4–C5–C8–C9–C10–C11–C14–P2–O4)2 (Fig. S3b, ESI†). The 2D double-layer structure is formed by π–π interactions (3.76 Å) between two central benzene rings of the organic ligands between layers A and B (Fig. 3).
 |
| | Fig. 1 ORTEP representation of a selected unit of compound 1. The thermal ellipsoids are shown at the 50% probability level. All H atoms are omitted for clarity. Symmetry code for the generated atoms: (A) x − 1, y + 1, z; (B) x − 1, y, z; (C) x − 2, y + 1, z. | |
 |
| | Fig. 2 The coordination modes of phosphonate ligands in compound 1 (a), compound 2 (b), and (c); coordination modes of the 1,4-benzenedicarboxylic acid ligands in compound 2 (d), (e), and (f). | |
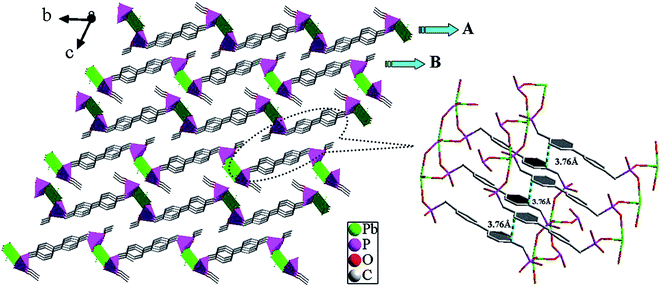 |
| | Fig. 3 The 2D double-layer structure of compound 1 via π–π stacking interactions; the π–π stacking interactions between the adjacent benzene rings with a face-to-face distance of 3.76 Å. | |
Crystal structures of [Pb3(BPDP)1.5(OOCC6H4COOH)3] (2). Single crystal X-ray diffraction analysis reveals that compound 2 also crystallizes in Triclinic P space group (see Table 1), having three independent Pb(II) ions with different coordination environments. As shown in Fig. 4, the Pb(1) center is coordinated to five O atoms: two O atoms (O1, O4B) from two BPDP2− anions and the extra O atom (O10, O11, and O17D) from two Hbdc− anions. The Pb(2) ion is four-coordinated by two O atoms (O14 and O16C) from two Hbdc− anions and two O atoms (O2, O7) from one BPDP2− anion to display a distorted tetrahedral geometry. The Pb(3) ion with a distorted tetrahedral geometry is coordinated with two O atoms (O18, O19) from one Hbdc− anion and two O atoms (O8 and O6A) from two BPDP2− anions. The Pb–O bond lengths are in the range 2.266(6) to 2.740(6) Å, which is comparable to those reported for other lead(II) phosphonates.17 As shown in Fig. 2b and c, the BPDP2− ligand has two types of coordination modes, and the Hbdc− anion displays three coordination modes in compound 2. All of the Hbdc− anions act as bidentate ligands (Fig. 2d–f). The {Pb(1)O5}, {Pb(2)O4}, and {Pb(3)O4} polyhedra and {CPO3} tetrahedra are interconnected via corner-sharing to form a 2D layer structure in the ac-plane (Fig. S4, ESI†). Neighboring double layers are bridged by the BPDP2− ligands, leading to a 3D framework structure with two types of channel system running along the c-axis (Fig. 5a). Channel A is formed by 22-membered rings composed of four Pb(II) ions and two Hbdc− anions. The dimensions of the channel are estimated to be [3.76 Å (C33–C39) × 12.14 Å (O17–O17)] based on structural data. Channel B is assembled by 48-atom rings [8.54 Å (C21–C21) × 14.82 Å (C42–C42)], and consists of four Pb(II) ions, two BPDP2− ligands, and two Hbdc− anions (Fig. 5b). The ester moieties of the phosphonate ligand are orientated into the channel space. In contrast to the packing structure of compound 1, compound 2 shows a novel framework structure with a higher dimensionality. This indicates that the introduction of the second metal linkers may be a good way to obtain multidimensional framework structures.
 |
| | Fig. 4 ORTEP representation of a selected unit of compound 2. The thermal ellipsoids are drawn at the 50% probability level. All H atoms are omitted for clarity. Symmetry code for the generated atoms: (A) −x + 1, −y + 2, −z; (B) −x + 2, −y + 1,−z; (C) x − 1, y, z; (D) −x + 1, −y + 2, −z + 1. | |
 |
| | Fig. 5 (a) View of the 3D framework for compound 2 along the c-axis; (b) (A) the 22-membered rings in compound 2; (B) the 48-atom rings in compound 2. | |
IR spectroscopy
The IR spectra for compounds 1 and 2 are recorded in the region 4000–400 cm−1 (Fig. S5 and S6, ESI†). The absorption band observed at ∼3400 cm−1 can be assigned to the O–H stretching vibrations. The characteristic bands of the coordination of carboxylate groups are shown at about 1600 cm−1 for asymmetric vibrations and at about 1400 cm−1 for symmetric vibrations for compound 2.18 The P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–O– stretching and bending bands appear in the region of 1100–900 cm−1. Additional intense and sharp bands at low energy (between 850 and 400 cm−1) for compounds 1 and 2 are observed. These bands are probably due to bending vibrations of the tetrahedral {CPO3} groups19 and Pb–O stretching vibrations.
O and P–O– stretching and bending bands appear in the region of 1100–900 cm−1. Additional intense and sharp bands at low energy (between 850 and 400 cm−1) for compounds 1 and 2 are observed. These bands are probably due to bending vibrations of the tetrahedral {CPO3} groups19 and Pb–O stretching vibrations.
PXRD patterns and thermal analysis
In order to identify the stability of the whole frameworks, thermogravimetric (TG) analysis, derivative thermogravimetry (DTG) analysis, and powder X-ray diffraction (PXRD) measurements were performed. Thermal decomposition of the compounds was carried out at a heating rate of 10 °C min−1 under a static air atmosphere over the temperature range 50–1100 °C. The structural transformation is observed with TG curves, and supported by DTG studies. From the DTG curves of compound 1, it is shown that there are two peaks during the whole process (Fig. S7, ESI†). It is suggested that the two peaks respectively correspond to two combustion stages in the whole combustion process: the partial decomposition of the 4,4′-biphenyldiphosphonate(monoethyl ester) molecule (233–335 °C) and further decomposition of the organic phosphonate ligands (420–800 °C). The mass losses of the two stages are 12.8% and 25.6%, respectively. The total weight loss at 900 °C is 38.4%, and the residue was not characterized because it was amorphous. The TG curve of compound 1 indicates that this has higher thermal stability. Powder X-ray diffraction studies (PXRD) were performed from 25 to 240 °C. The powder XRD patterns demonstrate that the structure of compound 1 was thermally stable below 240 °C (Fig. S9a, ESI†). As seen from Fig. S8,† the combustion process of compound 2 consists of three main steps: the partial decomposition of the two H2bdc molecules (212–339 °C), the elimination of the organic groups and the collapse of the structures (339–459 °C), and further decomposition of the compound (459–580 °C). Also, from the results, it can be seen that the mass losses of the three stages were 19.3%, 13.6%, and 11.4%, respectively. The total weight loss of 44.3% is close to the calculated value (43.09%) if the final products are assumed to be Pb(PO3)2 and Pb2P2O7 in a 1:2 molar ratio. We attempted to confirm this supposition by PXRD, but the final residues are unidentified because they are amorphous. To further understand the thermal stability of compound 2, powder X-ray diffraction studies (PXRD) were performed from 25 to 220 °C. In the end, the powder XRD patterns demonstrated the retention of the framework structure of compound 2 below 220 °C (Fig. S9b, ESI†).
Luminescence properties
The solid-state emission spectra of compounds 1 and 2 were recorded at room temperature. As shown in Fig. S10,† the free H2BPDP ligand shows an emission band at 422 nm (λex = 370 nm). The H2bdc ligand displays a fluorescent emission band at λmax = 398 nm with a relatively strong wavelength under the same excitation (Fig. S10b, ESI†). Upon excitation of 1 and 2 at 350 nm, the maximum emission peaks of the compounds are observed at 425 nm and 423 nm, respectively. Compared with that of the free H2BPDP ligand, the emission intensities of both compounds are much stronger. These emission bands are neither metal-to-ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT) in nature, but rather can be attributed to an intraligand emission state, because their fluorescence emission bands are very similar to that of the free ligand.20 The luminescence behaviors of compounds 1 and 2 are slightly different, and the PL quantum yields are 8% and 15% for compounds 1 and 2, respectively. This phenomenon reveals that the luminescence behavior is closely related to the channels of the compounds and the coordination modes of the ligands around the center Pb(II) ions. The investigation of luminescence properties shows that compounds 1 and 2 may be good blue-light luminescent materials. For further research, the luminescence emission of compounds 1 and 2 dispersed in different small molecule solvents, such as ethanol, methanol, 1-propanol, 1-butanol, 1-pentanol, N,N-dimethylformamide (DMF), and glycol, was investigated at room temperature (Fig. S11 and S12, ESI†). Among these solvents, ethanolic dispersions of compounds 1 and 2 revealed intense emission, so we chose ethanol as the solvent to investigate the luminescence properties of the compounds in ethanol suspension. As shown in Fig. S13,† compounds 1 and 2 have varying degrees of intense emission in ethanol. Therefore, the strong emissions of compounds 1 and 2 in the solid state and in organic suspensions confirm that they have potential applications in liquid-phase fluorescence detection.
Selective detection of nitroaromatic compounds
Compounds 1 and 2 are strongly luminescent in both the solid and liquid states, so we have studied their potential as a fluorescent probe for the selective and sensitive detection of nitroaromatic compounds (NACs). For luminescence sensing studies, the following NACs (1 mM in ethanol) were chosen: nitromethane (NM), m-dinitrobenzene (m-DNB), nitrobenzene (NB), o-nitrophenol (o-NP), p-nitrophenol (p-NP), 4-nitrotoluene (4-NT), and 2,6-dinitrotoluene (2,6-DNT). The suspensions were prepared by introducing 1.00 mg of powdered compounds 1 and 2 into 4.00 mL of ethanol. All of the titration experiments were executed by gradually adding suspension solutions in an incremental mode. The fluorescence quenching of compounds 1 and 2 is significantly affected by gradual addition of p-NP, and the other six analytes produce no obvious changes (Fig. S14–S27, ESI†). As depicted in Fig. 6a and b, at a concentration of 60 mM (240 μL), the quenching efficiency of compound 1 was estimated to be 85%, 66%, 55%, 54%, 52%, 20%, and 4% for p-NP, 4-NT, NB, 2,6-DNT, o-NP, m-DNB, and NM, respectively [quenching percentage = (I0 − I)/I0 × 100%, where I0 and I are the fluorescence intensities of compounds before and after addition of NACs, respectively]. As shown in Fig. 6c, although all the analytes could weaken the fluorescence intensity of compound 2, the fluorescence quenching percentage was quite different. According to the equation, the results of quenching are (from the highest to the lowest): p-NP > 4-NT > o-NP > m-DNB > NB = 2,6-DNT > NM. Moreover, the quenching percentage for p-NP reached 76% when the added quantity of analytes was 280 μL (70 mM) (Fig. 6d). The structures of the six analytes all have benzene rings, which can form π–π stacking interactions with the benzene rings in the ligands of compounds 1 and 2. Compared with the six analytes, NM has no aromatic rings, and the fluorescence quenching effect of NM was weaker than that of the other six analytes. The p-NP and o-NP contain hydroxyl groups that can act as hydrogen-bonding interaction sites, while other analytes (such as 4-NT, m-DNB, NB, and 2,6-DNT) have no hydroxyl groups, so the quenching of the compounds for these analytes is not obvious. However, the o-NP exhibits intramolecular hydrogen-bonding interactions (Fig. S28, ESI†), although the quenching ability of o-NP is poor. In summary, compounds 1 and 2 are highly selective and sensitive for p-NP.
 |
| | Fig. 6 (a, c) The fluorescence quenching experiments for compounds 1 and 2 dispersed in ethanol with addition of NACs, respectively; (b, d) percentage of fluorescence quenching resulting from the introduction of various NACs into EtOH-emulsions of compounds 1 and 2 at 60 mM and 70 mM concentrations, respectively. | |
Furthermore, to test the sensitivity of compounds 1 and 2 for p-NP, the emission spectra were recorded with addition of increasing concentrations of p-NP. As shown in Fig. S19a,† the fluorescence intensity of compound 1 reduces gradually with an increasing concentration of p-NP (10 mM to 60 mM). And compound 2 also revealed similar sensing behavior to p-NP (10 mM to 70 mM) (Fig. S30a†). These results indicated that compounds 1 and 2 have a high sensitivity of fluorescence quenching. Furthermore, the quenching efficiencies of compounds 1 and 2 were evaluated based on the Stern–Volmer (SV) equation:21 I0/I = 1 + Ksv[M], where [M] is the molar concentration of the NACs and Ksv is the quenching coefficient (unit: M−1). It is noteworthy that the S–V plots for p-NP are almost linear at low concentrations and bend upwards at higher concentrations of compounds 1 and 2 (Fig. S31a and S31b†). The quenching coefficients for p-NP are found to be 6.45 × 104 M−1 and 4.20 × 104 M−1 for compounds 1 and 2, respectively (Fig. S29b and S30b†). PXRD patterns of samples of compounds 1 and 2 before and after immersion in p-NP solution were obtained (Fig. S45a and S46a, ESI†). The diffraction peaks on the patterns correspond well with respect to position, demonstrating that the frameworks of compounds 1 and 2 remain complete even after immersion in p-NP solution. In order to study the recyclability of the compounds, recycling experiments with p-NP were carried out. The used samples were recovered by simple centrifugation and washing several times with ethanol. After five cycles, the initial luminescence intensities of the two compounds did not change significantly (Fig. S32†). This indicates the high retrievability of compounds 1 and 2.
Selective detection of Fe3+ ions
The potential application of compounds 1 and 2 for detecting metal ions was investigated in liquid suspensions: their crystalline samples were simply soaked in aqueous solutions of MClx (3.00 mL, 0.01 mol L−1, M = Zn2+, Al3+, Pb2+, Sr2+, Li+, Mg2+, Na+, Ba2+, Ca2+, Mn2+, Cd2+, Ni2+, Co2+, Fe2+, Cr3+, Cu2+, and Fe3+) for 12 h at room temperature to give metal-ion-incorporated suspensions of the compounds (Mn+@1 and Mn+@2) for luminescence measurements. The emission intensities of the different suspensions depend strongly on the species of metal ion (Fig. S34a† and 7a). As shown in Fig. S34b† and 7b, the presence of Fe3+ aqueous solution almost completely quenched the luminescence intensity of compounds 1 and 2 for the given metal ions, reducing it by approximately 100% compared with that of the original compounds 1 and 2 in water [quenching percentage = (I0 − I)/I0 × 100%, where I0 and I are the fluorescence intensities of compounds 1 and 2 before and after addition of metal ions, separately], whereas others enhanced or decreased the luminescence intensity to a degree. Moreover, under irradiation with 365 nm UV light, the Mn+@1 and Mn+@2 solutions all exhibited a blue color, which could be observed by the naked eye, but Fe3+@1 and Fe3+@2 displayed a dark color (Fig. S33a and b, ESI†). As can be seen, the emission intensity of samples of compounds 1 and 2 decreased as the concentration of Fe3+ (10 to 100 μM) was increased (Fig. S35c and S36c†). In order to further investigate the correlation between quenching and the Fe3+ concentration, as depicted in Fig. S35d and S36d,† a linear fluorescence intensity vs. Fe3+ concentration plot was made, which can be matched through the equation: I0/I = Ksv[Fe3+] + 1 (Ksv is the quenching rate constant and [Fe3+] is the molar concentration of Fe3+). The Stern–Volmer (SV) quenching constant (Ksv) was estimated from the plot of relative fluorescence intensity (I0/I) against the concentration of Fe3+. The dynamic quenching constants are calculated as 2.2 × 104 M−1 for compound 1 and 2.23 × 104 M−1 for compound 2.
 |
| | Fig. 7 (a) Luminescence intensity of compound 2 treated with 1.0 × 10−3 M various cations for 12 h; (b) percentage of fluorescence quenching as a result of introducing aqueous solutions of different metal cations. | |
In addition, in order to study the influence of different solvents on the luminescence of compounds 1 and 2, the as-synthesized samples (3.00 mg) were dispersed in 3.00 mL of 0.01 M ethanol suspensions of metal ions: MClx (M = Zn2+, Ba2+, Sr2+, Co2+, Na+, Mg2+, Pb2+, Ca2+, Al3+, Cd2+, Li+, Mn2+, Cr3+, Ni2+, Cu2+, Fe2+, and Fe3+) to form suspensions. The results show that some of the metal ions (Zn2+, Mg2+, Li+, and Ca2+) can enhance the fluorescence intensity of compounds 1 and 2, and the others exhibit different fluorescence quenching efficiencies (Fig. S37a and S38a, ESI†). And the quenching efficiency of compound 1 was calculated to be 97.4, 97.5, 99.2, 99.2, and 99.5% for Cr3+, Ni2+, Cu2+, Fe2+, and Fe3+ (Fig. S37b, ESI†). For compound 2, it is estimated that the quenching efficiency is 87, 90, 90, 91.2, 92.4, 92.4, 93.9, 96.3, 96.9, 98.5, and 100% for Al3+, Na+, Pb2+, Zn2+, Ni2+, Mn2+, Co2+, Cr3+, Cu2+, Fe2+, and Fe3+ (Fig. S38b, ESI†), respectively. For the Fe3+ ion, when the concentration of Fe3+ was increased, the emission intensities of the samples gradually decreased (Fig. S37c and S38c, ESI†). As shown in Fig. S37d and S38d,† the quenching coefficients for Fe3+ are found to be 1.77 × 104 M−1 and 6.0 × 103 M−1 for compounds 1 and 2. These results indicated that compounds 1 and 2 can detect more than one kind of metal ion in ethanol solution, but can detect only one kind of metal ion in aqueous solution. This indicates that the different solvents have different effects on ion recognition by compounds 1 and 2. The luminescence emissions of compounds 1 and 2 dispersed in water and ethanol solutions were investigated. In these two kinds of solvents, the fluorescence intensities of compounds 1 and 2 are stronger in water than in ethanol solvent (Fig. S39, ESI†). In a word, the luminescence experiments revealed that compounds 1 and 2 can be highly effective and selective luminescent sensors for Fe3+ ions in aqueous solutions.
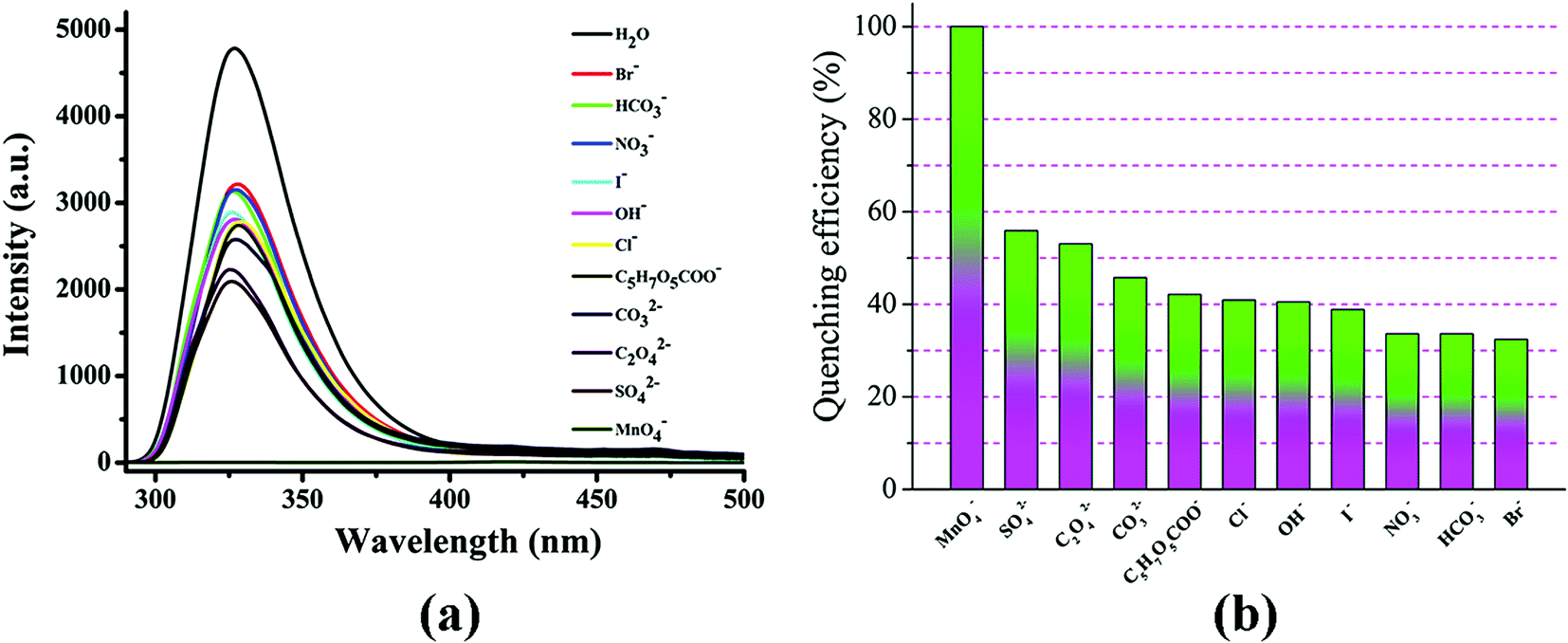
Selective detection of MnO4− ions
Crystalline samples of compounds 1 and 2 (3.00 mg) were added as a suspension into aqueous solutions, containing different potassium salt solutions of the same concentration (10−3 M, 3.00 mL) of Br−, HCO3−, NO3−, I−, OH−, Cl−, C5H7O5COO−, CO32−, C2O42−, SO42−, and MnO4−. As depicted in Fig. 8a, only MnO4− anions exhibited a significant fluorescence quenching effect, while the other anions did not cause a significant change in the fluorescence intensity of compound 1. For compound 2, the presence of Br−, NO3−, OH−, SO42−, HCO3−, C2O42−, CO32−, I−, and C5H7O5COO− ions in an aqueous solution decreases the luminescence intensity, but the effect was not as obvious as that of the MnO4− ion (Fig. 9a). As shown in Fig. 8b and 9b, MnO4− was also the most effective quencher and resulted in 100% quenching efficiency of compounds 1 and 2 (quenching percentage = (I0 − I)/I0 × 100%, where I0 is the initial emission peak intensity, I is the emission peak intensity with added MnO4− ions of molar concentration [MnO4−]). This result indicates that compounds 1 and 2 have highly selective detection and specific recognition of MnO4− anions in aqueous solution. It can be seen that the emission intensity of samples of 1 and 2 decreased with the concentration of MnO4− (10 to 100 μM) (Fig. S40c and S41c†). In order to further investigate the correlation between quenching and the MnO4− concentration, as depicted in Fig. S40d and S41d,† a linear fluorescence intensity vs. MnO4− concentration plot was made, which can be matched through the Stern–Volmer (SV) equation: I0/I = Ksv[MnO4−] + 1 (Ksv is the quenching constant and [MnO4−] is the concentration of the quencher). The Stern–Volmer (SV) quenching constant (Ksv) was estimated from the plot of relative fluorescence intensity (I0/I) against the concentration of MnO4−. The quenching constants (Ksv) of compounds 1 and 2 for MnO4− are calculated as 9.71 × 104 M−1 and 1.1 × 104 M−1, respectively.
 |
| | Fig. 8 (a) Luminescence intensity of compound 1 treated with 1.0 × 10−3 M various anions for 12 h; (b) percentage of fluorescence quenching as a result of introducing aqueous solutions of different anions. | |
 |
| | Fig. 9 (a) Luminescence intensity of compound 2 treated with 1.0 × 10−3 M various anions for 12 h; (b) percentage of fluorescence quenching as a result of introducing aqueous solutions of different anions. | |
Under the same measurement, suspensions were prepared by introducing 3.00 mg of powdered compounds 1 and 2 into ethanol as standard suspensions. For compound 1, MnO4− exhibits the highest fluorescence quenching of the emission intensity (Fig. S42a, ESI†), as is the case for compound 2 (Fig. S43a, ESI†). As depicted in Fig. S41b,† the quenching efficiency histograms show that I−, SO42−, C5H7O5COO−, HCO3−, C2O42−, Cl−, NO3−, and Br− caused larger but similar quenching intensity changes in compound 2. Furthermore, we examined the sensing of MnO4− in detail; as shown in Fig. S42c and S43c,† the luminescence intensities of compounds 1 and 2 in the suspensions gradually decreased with the addition of MnO4−. The quenching coefficients for MnO4− ions are calculated as 2.98 × 104 M−1 and 5.17 × 103 M−1 for compounds 1 and 2, respectively (Fig. S42d and S43d, ESI†). Therefore, compounds 1 and 2 can be employed as excellent candidate materials for selective sensing of MnO4− anions. Recycling experiments with compounds 1 and 2 have been performed. The used samples (8 mg) were recovered via simple centrifugation and washing several times with aqueous solution. The initial fluorescence intensities and quenching abilities of the compounds changed slightly over five repeated cycles (Fig. S44, ESI†). It is confirmed that compounds 1 and 2 have excellent recovery.
Possible detection mechanisms for ions
PXRD patterns of samples of compounds 1 and 2 before and after immersion in Fe3+ and MnO4− aqueous solution were obtained (Fig. S45 and S46, ESI†). The diffraction peaks on the patterns correspond well, demonstrating that the frameworks of compounds 1 and 2 remained complete even after immersion in Fe3+ and MnO4− aqueous solution. The quenched reason was not the collapse of the crystal structure caused by the ions. Furthermore, the UV-vis spectra of Fe3+ and MnO4− in aqueous solutions show strong absorption bands from 250 to 448 nm and 270 to 440 nm, respectively. For compounds 1 and 2, the strong absorption bands are also in the range 270–354 nm (Fig. S47†). Obviously, the absorption bands of compounds 1 and 2 are completely mantled by those of Fe3+ and MnO4− in aqueous solutions, resulting in a drastic decrease in luminescence intensities and even quenching.22 In addition, solid samples of compounds 1 and 2 were soaked in Fe3+ and MnO4− aqueous solutions, and then repeatedly washed with water. It was found that the color of the powder changed from white to yellow and gray, respectively (Fig. S48 and S49, ESI†), which confirmed that Fe3+ and MnO4− are diffused into the frameworks of compounds 1 and 2. A possible reason for the quenching mechanism is the electronic interaction between the metal ions and organic frameworks.23 The inductively coupled plasma spectroscopy (ICP) result showed that compounds 1 and 2 contained trace amounts of Fe and Mn, which were detected in the filtrate after immersion for several days, revealing the interaction between the ions and frameworks (Table S3†). Compound 2 has uncoordinated carboxyl oxygen atoms, which act as electron donors, and Fe3+ acts as an electron acceptor to form an electron-deficient area. This leads to energy migration. So, the quenching mechanism could involve donor–acceptor electron transfer. Other possible sensing mechanisms for luminescence quenching by Fe3+ ions needed to be further invested. As far as we all know, work on metal phosphonates as fluorescent probes for discriminating and sensitively detecting Fe3+ and MnO4− ions are still at a primary stage. Therefore, further studies will focus on the synthesis of novel fluorescent metal phosphonates and their application in the detection of ions.
Conclusions
In this paper, by using 4,4′-biphenyldiphosphonate(monoethyl ester) (H2BPDP) as the phosphonate ligand and 1,4-benzenedicarboxylic acid as the second metal linker, two novel lead(II) phosphonates with a 2D double-layer and a 3D framework structure have been successfully synthesized. The solid-state fluorescence properties of compounds 1 and 2 indicate that they are good candidates for blue-light luminescent materials. Importantly, compounds 1 and 2 show good sensing capabilities for nitroaromatics (NACs) and ions, especially p-NP, Fe3+, and MnO4− ions, by a fluorescence technique. The quenching mechanisms with these might involve the combined processes of electrostatic interactions between p-NP and organic ligands, and electron-transfer between the metal ions and organic frameworks. Research on other metal phosphonates for selective sensing of NACs, metal cations, and anions is currently underway in our group.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21371085).
Notes and references
-
(a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed;
(b) J. W. Ye, L. M. Zhao, R. F. Bogale, Y. Gao, X. X. Wang, X. M. Qian, S. Guo, J. Z. Zhao and G. L. Ning, Chem.–Eur. J., 2015, 21, 2029–2037 CrossRef CAS PubMed;
(c) S. K. Mostakim and S. Biswas, CrystEngComm, 2016, 18, 3104–3113 RSC;
(d) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed;
(e) M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef PubMed;
(f) C. S. Cao, H. C. Hu, H. Xu, W. Z. Qiao and B. Zhao, CrystEngComm, 2016, 18, 4445–4451 RSC.
-
(a) L. Li, Q. Chen, Z. G. Niu, X. H. Zhou, T. Yang and W. Huang, J. Mater. Chem. C, 2016, 4, 1900–1905 RSC;
(b) S. S. Zhao, J. Yang, Y. Y. Liu and J. F. Ma, Inorg. Chem., 2016, 55, 2261–2273 CrossRef CAS PubMed;
(c) X. Q. Wang, L. L. Zhang, J. Yang, F. L. Liu, F. N. Dai, R. M. Wang and D. F. Sun, J. Mater. Chem. A, 2015, 3, 12777–12785 RSC;
(d) M. M. Chen, X. Zhou, H. X. Li, X. X. Yang and J. P. Lang, Cryst. Growth Des., 2015, 15, 2753–2760 CrossRef CAS;
(e) Z. Chen, Y. W. Sun, L. L. Zhang, D. Sun, F. L. Liu, Q. G. Meng, R. M. Wang and D. F. Sun, Chem. Commun., 2013, 49, 11557–11559 RSC;
(f) M. H. Zhang, L. L. Zhang, Z. Y. Xiao, Q. H. Zhang, R. M. Wang, F. N. Dai and D. F. Sun, Sci. Rep., 2016, 6, 20672 CrossRef CAS PubMed.
-
(a) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed;
(b) A. J. Lan, K. H. Li, H. H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. C. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed;
(c) D. Banerjee, Z. C. Hu and J. Li, Dalton Trans., 2014, 43, 10668–10685 RSC;
(d) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
-
(a) Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Costero, M. Parraad and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC;
(b) H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821–3830 CrossRef CAS PubMed.
-
(a) S. Ghosh and P. S. Mukherjee, Organometallics, 2008, 27, 316–319 CrossRef CAS;
(b) G. V. Zyryanov, M. A. Palacios and P. Anzenbacher Jr, Org. Lett., 2008, 10, 3681–3684 CrossRef CAS PubMed;
(c) B. Gole, S. Shanmugamraju, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 10046–10048 RSC;
(d) S. L. Jackson, A. Rananaware, C. Rix, S. V. Bhosale and K. Latham, Cryst. Growth Des., 2016, 16, 3067–3071 CrossRef CAS.
-
(a) B. Gole, A. K. Bar and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 2276–2291 CrossRef CAS PubMed;
(b) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881–2885 CrossRef CAS PubMed;
(c) D. X. Ma, B. Y. Li, X. J. Zhou, Q. Zhou, K. Liu, G. Zeng, G. H. Li, Z. Shi and S. H. Feng, Chem. Commun., 2013, 49, 8964–8966 RSC;
(d) S. Sanda, S. Parshamoni, S. Biswas and S. Konar, Chem. Commun., 2015, 51, 6576–6579 RSC.
- A. Barba-Bon, A. M. Costero, S. Gil, M. Parra, J. Soto, R. Martinez-Manez and F. Sancenon, Chem. Commun., 2012, 48, 3000–3002 RSC.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
-
(a) W. Y. Dan, X. F. Liu, M. L. Deng, Y. Ling, Z. X. Chen and Y. M. Zhou, Dalton Trans., 2015, 44, 3794–3800 RSC;
(b) H. B. Xu, S. H. Zhou, L. L. Xiao, H. H. Wang, S. Z. Li and Q. H. Yuan, J. Mater. Chem. C, 2015, 3, 291–297 RSC;
(c) J. Zhao, Y. N. Wang, W. W. Dong, Y. P. Wu, D. S. Li and Q. C. Zhang, Inorg. Chem., 2016, 55, 3265–3271 CrossRef CAS PubMed;
(d) Z. M. Hao, X. Z. Song, M. Zhu, X. Meng, S. N. Zhao, S. Q. Su, W. T. Yang, S. Y. Song and H. J. Zhang, J. Mater. Chem. A, 2013, 1, 11043–11050 RSC;
(e) N. V. Ghule, R. S. Bhosale, A. L. Puyad, S. V. Bhosale and S. V. Bhosale, Sens. Actuators, B, 2016, 227, 17–23 CrossRef CAS.
- B. Ding, S. X. Liu, Y. Cheng, C. Guo, X. X. Wu, J. H. Guo, Y. Y. Liu and Y. Li, Inorg. Chem., 2016, 55, 4391–4402 CrossRef CAS PubMed.
-
(a) M. Elavarasi, A. Rajeshwari, S. A. Alex, D. N. Kumar, N. Chandrasekaran and A. Mukherjee, Anal. Methods, 2014, 6, 5161–5167 RSC;
(b) L. L. Li, L. L. Fan, M. Sun, H. M. Qiu, X. J. Li, H. M. Duan and C. N. Luo, Colloids Surf., B, 2013, 107, 76–83 CrossRef CAS PubMed.
-
(a) H. Zhu, J. Huang, S. S. Bao, M. Ren and L. M. Zheng, Dalton Trans., 2013, 42, 14075–14080 RSC;
(b) S. F. Tang, L. J. Li, X. X. Lv, C. Wang and X. B. Zhao, CrystEngComm, 2014, 16, 7043–7052 RSC;
(c) L. H. Schilling and N. Stock, Dalton Trans., 2014, 43, 414–422 RSC;
(d) F. P. Zhai, Q. S. Zheng, Z. X. Chen, Y. Ling, X. F. Liu, L. H. Weng and Y. M. Zhou, CrystEngComm, 2013, 15, 2040–2043 RSC.
-
(a) L. Liu, J. Y. Hao, Y. T. Shi, J. S. Qiu and C. Hao, RSC Adv., 2015, 5, 3045–3053 RSC;
(b) J. Z. Wang, W. Sun, S. Y. Chang, H. T. Liu, G. N. Zhang, Y. Q. Wang and Z. L. Liu, RSC Adv., 2015, 5, 48574–48579 RSC;
(c) M. Venkateswarulu, A. Pramanik and R. R. Koner, Dalton Trans., 2015, 44, 6348–6352 RSC;
(d) W. J. Li, J. Lü, S. Y. Gao, Q. H. Li and R. Cao, J. Mater. Chem. A, 2014, 2, 19473–19478 RSC.
-
(a) S. P. Shi, Y. Y. Zhu, Z. G. Sun, W. Zhou, L. L. Dai, M. X. Ma, W. Z. Li, H. Luo and T. Sun, Cryst. Growth Des., 2014, 14, 1580–1590 CrossRef CAS;
(b) L. L. Dai, Y. Y. Zhu, C. Q. Jiao, Z. G. Sun, S. P. Shi, W. Zhou, W. Z. Li, T. Sun, H. Luo and M. X. Ma, CrystEngComm, 2014, 16, 5050–5061 RSC;
(c) C. Ma, C. Q. Jiao, Z. G. Sun, Y. Y. Zhu, X. W. Zhang, M. L. Wang, D. Yang, Z. Zhao, H. Y. Li and B. Xing, RSC Adv., 2015, 5, 79041–79049 RSC.
- J. W. Zhang, C. C. Zhao, Y. P. Zhao, H. Q. Xu, Z. Y. Du and H. L. Jiang, CrystEngComm, 2014, 16, 6635–6644 RSC.
- G. M. Sheldrick, Acta Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
-
(a) C. Gabriel, C. P. Raptopoulou, A. Terzis, V. Psycharis, F. Gul-E-Noor, M. Bertmer, C. Mateescu and A. Salifoglou, Cryst. Growth Des., 2013, 13, 2573–2589 CrossRef CAS;
(b) C. Gabriel, P. Karakosta, A. A. Vangelis, C. P. Raptopoulou, A. Terzis, V. Psycharis, M. Bertmer, C. Mateescu and A. Salifoglou, Cryst. Growth Des., 2015, 15, 1666–1682 CrossRef CAS.
-
(a) M. Bazaga-García, M. Papadaki, R. M. P. Colodrero, P. Olivera-Pastor, E. R. Losilla, B. Nieto-Ortega, M. A. G. Aranda, D. Choquesillo-Lazarte, A. Cabeza and K. D. Demadis, Chem. Mater., 2015, 27, 424–435 CrossRef;
(b) Z. S. Cai, S. S. Bao, X. Z. Wang, Z. Hu and L. M. Zheng, Inorg. Chem., 2016, 55, 3706–3712 CrossRef CAS PubMed;
(c) X. B. Liu, H. Lin, Z. Y. Xiao, W. D. Fan, A. Huang, R. M. Wang, L. L. Zhang and D. F. Sun, Dalton Trans., 2016, 45, 3743–3749 RSC.
-
(a) R. B. Fu, S. M. Hu and X. T. Wu, CrystEngComm, 2013, 15, 8937–8940 RSC;
(b) R. Silbernagel, C. H. Martin and A. Clearfield, Inorg. Chem., 2016, 55, 1651–1656 CrossRef CAS PubMed;
(c) M. Taddei, P. Sassi, F. Costantino and R. Vivani, Inorg. Chem., 2016, 55, 6278–6285 CrossRef CAS PubMed.
-
(a) X. D. Zhu, Y. Li, W. X. Zhou, R. M. Liu, Y. J. Ding, J. Lü and D. M. Proserpio, CrystEngComm, 2016, 18, 4530–4537 RSC;
(b) X. B. Liu, Z. Y. Xiao, A. Huang, W. Wang, L. L. Zhang, R. M. Wang and D. F. Sun, New J. Chem., 2016, 40, 5957–5965 RSC;
(c) A. Buragohain, M. Yousufuddin, M. Sarma and S. Biswas, Cryst. Growth Des., 2016, 16, 842–851 CrossRef CAS;
(d) H. Zhang, J. Yang, Y. Y. Liu, S. Y. Song and J. F. Ma, Cryst. Growth Des., 2016, 16, 3244–3255 CrossRef CAS.
-
(a) C. Q. Zhang, Y. Yan, L. B. Sun, Z. Q. Liang and J. Y. Li, CrystEngComm, 2016, 18, 4102–4108 RSC;
(b) R. B. Lin, F. Li, S. Y. Liu, X. L. Qi, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2013, 52, 13429–13433 CrossRef CAS PubMed.
-
(a) G. P. Li, G. Liu, Y. Z. Li, L. Hou, Y. Y. Wang and Z. H. Zhu, Inorg. Chem., 2016, 55, 3952–3959 CrossRef CAS PubMed;
(b) Y. T. Liang, G. P. Yang, B. Liu, Y. T. Yan, Z. P. Xia and Y. Y. Wang, Dalton Trans., 2015, 45, 13325–13330 RSC;
(c) J. M. Zhou, W. Shi, H. M. Li, H. Li and P. J. Cheng, J. Phys. Chem. C, 2014, 118, 416–426 CrossRef CAS.
- Y. L. Wu, G. P. Yang, Y. D. Zhang, N. N. Shi, J. Han and Y. Y. Wang, RSC Adv., 2015, 5, 90772–90777 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format for compounds 1 and 2. XRD patterns, IR spectra, TGA curves, fluorescence spectra, 2D layer structure, the fluorescence properties of standard emulsions of compound 1 and compound 2 in the presence of various contents of NACs, reproducibility of the quenching efficiency of compounds after five recycles for p-NP, Fe3+, and MnO4−, XRD patterns of 1 and 2 dispersed in p-NP, Fe3+, and MnO4− solutions, ICP, UV-vis spectra, UV light irradiation images of compounds in ethanol solvents in a series of ion recognition pictures. Tables S1 and S2: selected bond lengths [Å] and angles for compounds 1 and 2. CCDC 1495968 (1) and 1495969 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra21403g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Dan Yang and
Jing Li
*,
Dan Yang and
Jing Li