
Arsenic speciation analysis in environmental water, sediment and soil samples by magnetic ionic liquid-based air-assisted liquid–liquid microextraction
Abstract
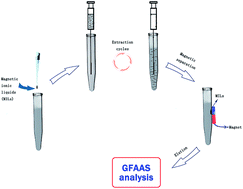
A novel magnetic ionic liquid-based air-assisted liquid–liquid microextraction (MIL–AALLME) technique was developed for highly selective separation of trace amounts of arsenite and arsenate species in various environmental water, sediment and soil samples prior to its determination by graphite furnace atomic absorption spectrometry (GFAAS). This highly efficient separation method combines the advantages of magnetic ionic liquid (MIL) and air-assisted liquid–liquid microextraction (AALLME) for the first time. The MIL, 1-butyl-3-methylimidazolium tetrachloroferrate ([C4mim][FeCl4]), was used as the extractant to simplify the microextraction procedure by magnetic separation. The variables of interest in the MIL–AALLME method, such as pH values, amounts of chelating agent, types and amounts of MIL, salt effect and number of extraction cycles were assessed and optimized systematically. Under optimal extraction conditions, the MIL–AALLME method provided a good linear dynamic range (LDR) in the range of 0.04–10.0 μg L−1 and the determination coefficient was 0.9991. Using the present method, the limit of detection (LOD) and the relative standard deviation (RSD) for seven replicate measurements of 2.0 μg L−1 of As(III) was 0.029 μg L−1 and 2.5%, respectively. The developed methodology was successfully used for inorganic arsenic speciation studies in different environmental water, sediment and soil samples with satisfactory results ranging from 93.0 to 108.5% for the spiked samples. In order to confirm the accuracy of MIL–AALLME, five standard reference materials such as GBW08605 simulated natural water and GBW07309 sediment were analyzed, and the analyzed values found by using the present methodology were in statistic agreement with the certified values.
 Please wait while we load your content...
Please wait while we load your content...

